A hydrogen bond occurs when two electronegative atoms, such as nitrogen
and oxygen, interact with the same hydrogen. The hydrogen is normally covalently
attached to one atom, the donor, but interacts electrostatically
with the other, the acceptor. This interaction is due to the dipole
between the electronegative atoms and the proton.
![]() Link here to discussion of H-bonds from
the PPS course.
Link here to discussion of H-bonds from
the PPS course.
There is a geometric component involved in hydrogen bonds, and for single donor acceptor systems, such as N-H---O, the strongest hydrogen bonds are collinear (Creighton, 1993 and references therein). Electrostatic calculations suggest that deviation of 20° from linearity leads to a decrease in binding energy of approximately 10% (Pimentel & McClellan, 1960).
In double acceptor systems, bifurcated hydrogen bonds with non-linear angles are preferred. The occurrence of hydrogen bonds in protein structure has been extensively reviewed by Baker & Hubbard (1984), albeit before the pdb database was as large as it is today. They found that 90% of N-H---O bonds in proteins lie between 140 and 180°, and that they are centred around 158°C. For C=O---H, the range is more broadly distributed between 90° and 160° and centred around 129°.
The groups that participate in hydrogen bond interactions in proteins
are shown here.![]() from the PPS course.
from the PPS course.
The strength of a hydrogen bond is between 2 and 10 kcal/mol, and one might think that this is the amount of energy one hydrogen bond contributes towards stabilization of a folded protein. However, in the unfolded state, all potential hydrogen bonding partners in the extended polypeptide chain are satisfied by hydrogen bonds to water. When the protein folds, these protein-to-water H-bonds are broken, and only some are replaced by (often sub-optimal) intra-protein H-bonds. McDonald & Thornton (1994) showed that while only 1.3% of backbone amino groups and 1.8% of carbonyl groups in proteins fail to H-bond (without any obviously compensating interactions), 80% of main chain carbonyls fail to form a second hydrogen bond. Thus, if one considers enthalpy terms alone, it would appear that hydrogen bonding is destabilizing to folded protein structure.
However, one must also consider entropy. When a protein folds, and those hydrogen bonds that the protein made to bulk water are broken, the entropy of the solvent increases. The balance between the entropy and enthalpy terms are close, and in the recent past it was considered that H-bonds made no contribution overall to protein stability. But, it is now generally accepted that H-bonds make a positive contribution to protein stabilisation (reviewed in Pace et al., 1996).
Estimation of the contribution of hydrogen bonding to protein stability
has been made by a combination of experiments on model compounds and site-directed
mutational (SDM) studies. The difficulty with the SDM studies is that when
a smaller residue replaces a larger one, a cavity is created. For example,
mutating Asn to Ala creates a cavity of 37.4 Å3 (Harpaz et al , 1994). This cavity
may then be filled by water, replacing the hydrogen bonding of the asparagine
NH group. Even in more conservative mutations such as Thr to Val (which
is isosteric) or Ser to Ala, one must take into account the contribution
of side chain entropy and the hydrophobic effect to the ![]() (
(![]() G) values. Dissection
of the latter contributions from those due only to hydrogen bonds is not
trivial. An estimation has been made of a positive contribution of 1.5±1.0
kcal/mol (Pace et al.,
1996, Fersht, 1987) from
the formation of a buried intramolecular uncharged hydrogen bond. However,
in order to form, the unfavourable interaction energy from burial of a polar
group must be overcome. Thus, the net energy gain for formation of a buried
H-bond is approximately 0.6 kcal/mol.
G) values. Dissection
of the latter contributions from those due only to hydrogen bonds is not
trivial. An estimation has been made of a positive contribution of 1.5±1.0
kcal/mol (Pace et al.,
1996, Fersht, 1987) from
the formation of a buried intramolecular uncharged hydrogen bond. However,
in order to form, the unfavourable interaction energy from burial of a polar
group must be overcome. Thus, the net energy gain for formation of a buried
H-bond is approximately 0.6 kcal/mol.
Despite the small contribution made to protein stability by hydrogen bonds, we must remember that if we break or delete an intramolecular hydrogen bond in a protein without the possibility of forming a compensating H-bond to solvent, that protein will be destabilized. In globular proteins, much of the H-bonding potential of the backbone amide and carbonyl groups is satisfied by the formation of regular structure such as alpha helix and beta sheet (links to PPS); regular structure comprises 80 - 90% of globular protein structure.
There is evidence that hydrogen bonds contribute to stability in hyper-thermostable proteins. A comparison of glyceraldehyde-3-phosphate dehydrogenase (GADH) from four organisms with a range of thermostabilities and more than 50% sequence identity found that the strongest correlation to thermostability was with the number of buried charged residues H-bonded to buried neutral residues (Tanner et al, 1996). The rationalization given for this preference of charged-to-neutral over neutral-to-neutral or charged-to-charged residue H-bonding was as follows: The enthalpy of H-bond formation is in the order, charged-to-charged > charged-to-neutral > neutral-to-neutral, but the entropic cost of desolvation is in the inverse order. The greatest overall free energy benefit is proposed to be for the charged-to-neutral H-bonds. If you wish to make this comparison yourself, here are links to the the GADH structures from Lobster [Bbk|BNL|ExP|Waw|Hal], Bacillus stearohermophilus [Bbk|BNL|ExP|Waw|Hal], Thermotoga maritima [Bbk|BNL|ExP|Waw|Hal] and Thermus aquaticus [Bbk|BNL|ExP|Waw|Hal].
Charged-to-charged residue H-bonds are a special case known as "Salt-Bridges" and their benefit-to-cost ratio is discussed below.
We can look at the effect of deleting H-bonds from a protein using site-directed mutagenesis. However, we must remember that these substitutions also alter non-H-bonding interactions.
Consider some examples from the ribonuclease barnase (Serrano et al, 1992).
The mutation of Tyr24 to Phe: Tyr 24 makes a hydrogen bond of good geometry
with the buried main chain carbonyl of Asp75. Deletion of this H-bond is
energetically neutral because the deletion
of the H-bond is balanced by the increase in hydrophobicity on mutating
Tyr to Phe.
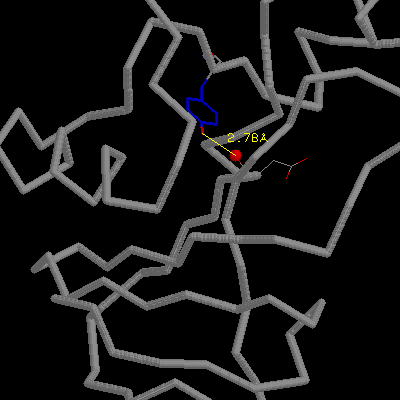 click here for gif and pdb
file.
click here for gif and pdb
file.
However, Tyr78 makes two H-bonds to Gly81; one to the buried main chain
carbonyl and one to the partially exposed amino group. Thus mutation Try78Phe
removes two H-bonds and leads to a net destabilization of 1.4 kcal/mol.
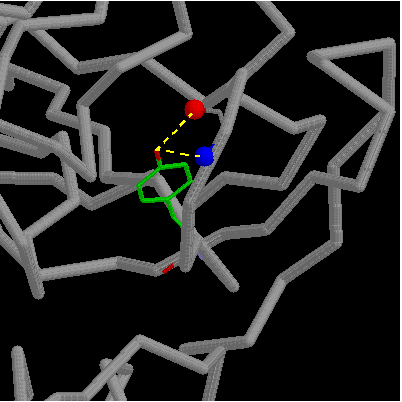 click here for gif and pdb
file.
click here for gif and pdb
file.
The OH of Ser92 is almost completely buried and makes two H-bonds with
the main chain amino and carbonyl groups of Thr70. Mutation Ser92Ala removes
these two H-bonds and leads to a net destabilization of 2.8 kcal/mol.
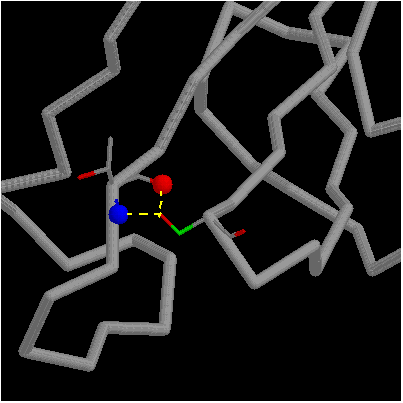 click here for gif and pdb
file.
click here for gif and pdb
file.
In the above cases, none of H-bond partners left behind after mutation had access to water. However, Ser57 makea single H-bond with a non-charged group exposed to solvent. Mutation of Ser to Ala leads to a net stabilization of 0.14 kcal/mol; this is presumably because the non-charged, solvent-exposed group makes better H-bonds to solvent than it did to serine57.
![]() The Hydrophobic Effect.
The Hydrophobic Effect. ![]() Conformational Entropy
Conformational Entropy![]() Beginning
Beginning