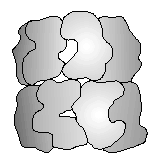
Fig. 1. The cpn60 14-mer. Subunit outlines based on the X-ray crystal structure of GroEL. Diagram adapted from Braig et al. (1994).
Helen R. Saibil
Department of Crystallography,
Birkbeck College London, Malet
St, London WC1E 7HX, UK
Published in 'The Chaperonins', RJ Ellis, Ed., Academic Press and reproduced by permission.
Fig. 1. The cpn60 14-mer. Subunit outlines based on the X-ray crystal structure of GroEL. Diagram adapted from Braig et al. (1994).
The EM methods for sample preparation and imaging will be briefly explained in the following sections. Very small amounts of protein are needed for EM study. Unlike the situation in optical microscopy, the depth of focus in transmission EM is much greater than the specimen thickness, so that the observed image is a projection through the whole structure. In order to get 3D information, images must be obtained of the specimen in different orientations so that the different views can be combined into a 3D density map. This is done by recording images with different tilt angles, and/or by taking advantage of molecular symmetry to generate other views. Transmission EM images are 2D projections of 3D structures, and 3D reconstruction can be done by computed tomography, if enough views at different angles are available.
In routine transmission EM, samples are imaged under high vacuum and therefore must be dehydrated. Protein structures can be imaged by allowing them to dry in a thin film of heavy metal stain, effectively encasing them in an electron dense medium. The image is formed by the stain, which reveals the surface shape of the protein by its exclusion. Although crude and simple to apply, negative staining often gives quite a good idea of the shape and symmetry of a protein structure to about 20 Å resolution. Its main drawbacks are variable distortion due to dehydration and lack of internal structure. Drying usually causes flattening of the 3D structure which poses particular problems for 3D reconstruction from negative stain images.
A technique increasingly used by structural biologists is cryo EM. The aqueous protein sample is applied to an EM grid, but instead of staining and dehydration, a thin layer of suspension is very rapidly cooled by plunging into a cold liquid (typically ethane, cooled to liquid nitrogen temperature). This traps the protein in the native, hydrated state, embedded in vitrified water. Rapid cooling prevents the formation of ice crystals, which would otherwise damage the protein by removing water from its surface, and the protein is preserved in the native state as long as the grid is kept below about -150/C (Dubochet et al., 1988). There are very significant advantages to this approach, mainly that the native protein electron density is imaged, and is directly comparable to that determined by X-ray crystallography, even though the resolution is normally much lower, of the order of 20-30 Å in single molecule work (as opposed to 2D crystal data which can go to atomic resolution). A disadvantage of cryo EM of unstained, frozen-hydrated proteins is the extremely low image contrast. This, combined with the need to limit the electron dose because of radiation damage, means that the signal to noise ratio is very low. In order to extract reliable information, it is necessary to average many copies of the protein image, by computer processing. This is essential in any case for 3D reconstruction, which requires the combination of many different views.
Crystallization and structure determination for a large protein with 7-fold symmetry is an enormous task. Highly purified protein must be available in large quantities to find good crystallization conditions and grow suitable crystals for data collection. Wild-type GroEL is difficult to crystallize, but Braig et al. (1994) found that a double mutant, Arg13®Gly, Ala126®Val formed good crystals. The C2221 crystal form contained one half molecule per asymmetric unit; a molecular 2-fold corresponded to a lattice 2-fold, reducing the problem of finding the molecule to that of positioning the 7-fold axis, known to be perpendicular to the 2-fold. Three heavy atoms per subunit were located in the heptameric ring to yield a 6 Å map by single isomorphous replacement; then 7-fold averaging was exploited to extend the phases from 6 to 2.8 Å. Because 7-fold symmetry does not fit into a crystal lattice, the 7 subunits make different packing interactions with neighbouring rings, and the more deformable parts of the structure deviate from exact 7-fold symmetry. Such regions are not resolved by methods that rely on 7-fold averaging. Most of the structure, with the notable exception of the termini and a region near the middle of the sequence, was clearly resolved by this procedure. Considerable work lies ahead, in the refinement of the structure and in the analysis of GroEL in complex with nucleotides, GroES and the various possible substrate combinations. All of these structures should illuminate the molecular mechanism of chaperonin-assisted protein folding.

Fig. 2. Cryo-EM averaged end and side views of GroEL (a,b), from Chen et al. (1994).
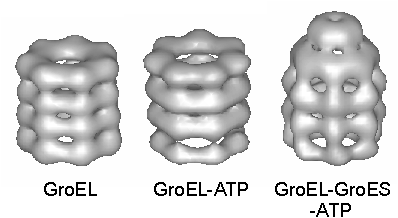
Fig. 3. Surface-rendered views of 3D reconstructions of GroEL, GroEL-ATP and GroEL-GroES-ATP from cryo-EM. The GroES ring is seen as a disk above the GroEL. Reprinted with permission from Nature (Chen et al., 1994). Copyright 1994 Macmillan Magazines Limited.
At the top of the equatorial domain, there is a well-defined junction with the intermediate domain, a small, antiparallel domain connecting the top and bottom large domains. It comprises a pair of crossed helices and a small J-strand region that contacts the neighbouring apical domain. This is followed by another junction leading to the large apical domain, which contains I and J structures and has poorly defined density in regions lining the channel, the site of residues involved in substrate and GroES binding, as identified by mutagenesis (see below). A conspicuous feature is that much of the apical domain surface is not in close contact with neighbouring domains, and that there is little barrier to its movement, both locally in some parts and in overall orientation of the domain. It is this part of the structure that deviates most from 7-fold symmetry in the crystal.
Very interesting potential hinge sites are found at the upper and lower domain junctions. These provide the possibility of large hinge movements allowing rigid body rotations (Gerstein et al., 1994) of the apical and equatorial domains. In these regions of exposed antiparallel chain, 3 of the 4 sequences contain conserved glycine residues. Fig. 6a shows a schematic diagram representing the subunit domain arrangement in the oligomer, in which the potential hinge sites are marked with black dots.

Fig. 4. Atomic resolution structure of the GroEL subunit (Braig et al., 1994). (a) alpha-carbon and (b) full structure, colour coded in rainbow colours from N (red) to C (blue). In this view, the equatorial domain is the large mass containing both termini at the bottom of the subunit, connected by the antiparallel intermediate domain (cyan and yellow) to the large apical domain (green and cyan) at the top. Images kindly provided by Dr. Kerstin Braig.
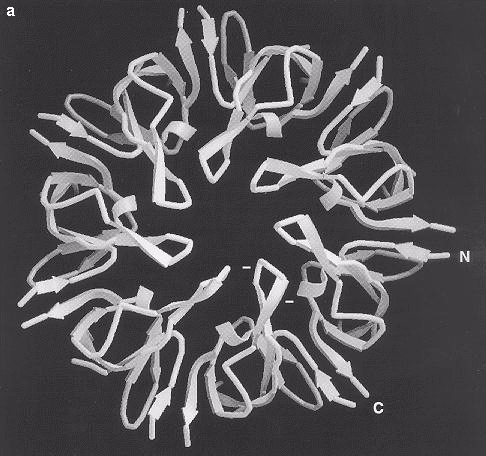
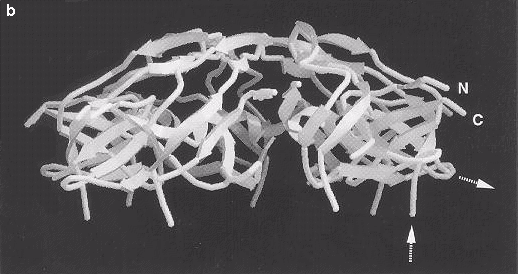
Fig. 5 Ribbon diagrams of the GroES structure viewed (a) from above and (b) from the side, showing the ß-barrel structure of the subunit with a ß-hairpin forming the roof of the dome-like heptamer. The N and C termini are labeled and point radially outward. In (a), the positions of two glutamic acid residues in the ß-hairpin are indicated as negative charges. The broken ends of the disordered mobile loop, which is not seen in this map, are indicated by dashed arrows in (b). Images kindly provided by Dr John Hunt.
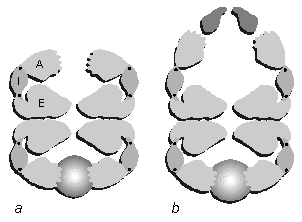
Fig. 6. Schematic diagram of the subunit arrangement in a hypothetical slice through the oligomer, showing the major functional sites. (a) GroEL, based on the crystal structure with bound substrate (shaded light to dark) as imaged by cryo-EM. A, apical domain; I, intermediate domain; E, equatorial domain. The apical domains form a ring of hand-like structures with the substrate binding sites on the fingers protruding into the central channel. Sites of potential hinge rotations are indicated by black dots. The notch in the equatorial domain represents the ATP-binding cleft. The interaction across the equatorial plane is shown as pairs of wavy surfaces. (b) The folding complex GroEL-MDH-GroES-ATP, rotating the subunit domains and adding the GroES (dark gray) and substrate (shaded) densities according to cryo EM observations.

Fig. 7. (a) Side view of the GroEL-GroES complex averaged from 200 cryo EM images (Chen et al., 1994). (b) Side view average of "football" complexes from cryo-EM (S. Chen, A. Roseman, and H. Saibil, unpublished observations). (c) Section through a 3D reconstruction of the complex in (a) showing the large reorientation of the apical domains in contact with GroES, forming the contact with the same region as the substrate binding site. The GroEL cylinder is greatly elongated. (d) Section of a 3D reconstruction of the folding complex GroEL-MDH-GroES-ATP trapped by vitrification after 12 s of folding from cryo-EM (Chen et al., 1994). The MDH substrate density is found in the opposite ring to GroES.
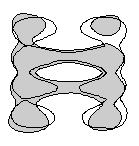
Fig. 8. Superposed outlines of sections through GroEL and GroEL-ATP (shaded) from cryo-EM (Chen et al., 1994), showing the slight but distinct outward rotation of the apical domains in the ATP form. These displacements have the effect of lengthening the cylinder and widening the binding cavity.
Averaged side views and sections through the GroEL-GroES complex shows large displacements of the apical domains, consistent with a large rotation of the apical domains in contact with GroES about the upper hinge region (Fig. 7a,c). This dramatic rotation occurs at both ends of GroEL in the case of the football structures (Fig 7b).
By cryo EM, the substrate malate dehydrogenase was directly observed as additional density at one end of GroEL, held between the apical domains, in the mouth of the central channel (Chen et al., 1994; diagrammed in Fig 6a). One-sided binding was observed even in the presence of greater than a 2-fold molar excess of MDH, implying a negative cooperativity of substrate binding. Because the vitrification procedure traps the sample rapidly from the native state, it is possible to capture transient complexes whose lifetime is longer than the mixing, blotting and freezing time (about 10 sec with normal procedures). The transient complex GroEL-GroES-ATP-MDH was captured and imaged within 12 sec of starting the folding reaction. It showed extra density, as compared to the 'empty' GroEL-GroES-ATP complex, in the apical cavity remote from the bound GroES (Chen et al., 1994; Fig 6b, 7c & d). Samples vitrified after longer periods of folding show a progressive emptying of the binding site (Roseman, A.M., Chen, S., Burston, S.G., Clarke, A.R. & Saibil, H.R., unpublished). The size and shape of the extra density are more suggestive of the native MDH subunit structure than of a highly expanded state.
In the crystal structure, Arg 197 in the apical domain is shown to be involved in a contact with Glu386 in the neighbouring intermediate domain. Mutation of R197®Ala reduces both the positive cooperativity of the ATPase within rings and its negative cooperativity between rings, leading to the proposal, now backed up by the crystal structure, that it is involved in both intra- and inter-subunit allosteric interactions (Yifrach and Horovitz, 1994). These residues are near the upper (apical) hinge, whereas the ATP-binding site is next to the lower hinge.
Other mutations, around the hinge regions, and at a site at the equatorial plane that forms a contact between the two rings, influence GroES binding. Glu461, which forms a charge pair contact between the two rings with Arg452, is particularly remote from the apical domain, and likely to be important in negative cooperativity.
A set of particularly interesting mutations point to allosteric sites affecting GroES binding and ATP hydrolysis. In addition to the sites that are likely to be in direct contact with these ligands, mutations of residues throughout the intermediate domain interfere with GroES binding and ATPase activity. The intermediate domain with its two potential hinges is thus heavily implicated in the allosteric interactions at all stages of the reaction cycle. Not surprisingly, mutations that affect GroES binding and ATP hydrolyis also affect polypeptide release and folding. Another line of communication, via a direct contact between apical and equatorial domains, is suggested by the effect of mutating Gly45, a residue in the equatorial domain that comes quite close to the apical domain. Mutating Gly45®Glu causes defective release without reducing ATPase or GroES binding. Intriguingly, other mutations with these effects are also found near the top of the apical domain (Glu238), and towards the bottom of the equatorial domain (Asp25). It appears that binding and release are blocked by very different mutations. Binding may be relatively straightforward to understand but release is complex, involving ATPase and GroES mechanisms. The wide distribution of sites affecting substrate release suggests that global rearrangements accompany the release step.
Azem, A., Kessel, M. and Goloubinoff, P. (1994). Characterization of a functional GroEL14(GroES7)2 chaperonin hetero-oligomer. Science 265, 653-656.
Baneyx, F. and Gatenby, A.A. (1992). A mutation in GroEL interferes with protein folding by reducing the rate of discharge of sequestered polypeptides. J. Biol. Chem. 267, 11637-11644.
Bochkareva, ES and Girshovich, AS (1994) ATP induces non identity of two rings in chaperonin GroEL. J. Biol. Chem. 269, 23869-23871.
Braig, K., Otwinowski, Z., Hegde, R., Boisvert, D.C., Joachimiak, A., Horwich, A.L. and Sigler, P.B. (1994). The crystal structure of the bacterial chaperonin GroEL at 2.8 Å. Nature 371, 578-586.
Braig, K., Simon, M., Furuya, F., Hainfeld, J.F. and Horwich, A.L. (1993). A polypeptide bound by the chaperonin groEL is localized within a central cavity. Proc. Natl. Acad. Sci. USA 90, 3978-3982.
Chandrasekhar, G.N., Tilly, K., Woolford, C., Hendrix, R. and Georgopoulos, C. (1986). Purification and properties of the groES morphogenetic protein of Escherichia coli. J. Biol. Chem. 261, 12414-12419.
Chen, S., Roseman, A.M., Hunter, A., Wood, S.P., Burston, S.G., Ranson, N., Clarke, A.R. and Saibil, H.R. (1994). Location of a folding protein and shape changes in GroEL-GroES complexes imaged by cryo-electron microscopy. Nature 371, 261-264.
Dubochet, J., Adrian, M., Chang, J.-J., Homo, J.-C., Lepault, J., McDowall, A.W. and Schultz, P. (1988). Cryo-electron microscopy of vitrified specimens. Quart. Rev. Biophys. 21, 129-228.
Fenton, W.A., Kashi, Y., Furtak, K. and Horwich, A.L. (1994). Residues in chaperonin GroEL required for polypeptide binding and release. Nature 371, 614-619.
Fisher, M.T. and Yuan, X. (1994). The rates of commitment to renaturation of rhodanese and glutamine synthetase in the presence of the GroE chaperonins. J. Biol. Chem.
Fisher, M. (1994). The effect of groES on the groEL-dependent assembly of dodecameric glutamine synthetase in the presence of ATP and ADP. J. Biol. Chem. 269, 13629-13636.
Gatenby, A.A. and Viitanen, P.V. (1994). Structural and functional aspects of chaperonin-mediated protein folding. Annu. Rev. Plant Physiol. Plant Mol. Biol. 45, 469-491.
Georgopoulos, C.P., Hendrix, R.W., Casjens, S.R. and Kaiser, A.D. (1973). Host participation in bacteriophage lambda head assembly. J. Mol. Biol. 76, 45-60.
Gerstein, M., Lesk, A.M. and Chothia, C. (1994). Structural mechanisms for domain movements in proteins. Biochemistry 33, 6739-6749.
Goloubinoff, P., Christeller, J.T., Gatenby, A.A. and Lorimer, G.H. (1989). Reconstitution of active dimeric ribulose bisphosphate carboxylase from an unfolded state depends on two chaperonin proteins and Mg-ATP. Nature 342, 884-889.
Harris, J.R., Zahn, R. and Plückthun, A. (1994). Transmission electron microscopy of GroEL, GroES and the symmetrical GroEL/ES complex. J. Struct. Biol. 112, 216-230.
Hartl, F.-U. and Martin, J. (1995). Molecular chaperones in cellular protein folding. Curr. Opinion in Struct. Biol. 5, 92-102.
Hendrix, R. (1979). Purification and properties of GroE, a host protein involved in bacteriophage assembly. J. Mol.Biol. 129, 375-392.
Hohn, T., Hohn, B., Engel, A., Wurtz, M. and Smith, P.R. (1979). Isolation and characterization of the host protein GroE involved in bacteriophage lambda assembly. J. Mol.Biol. 129, 359-373.
Horovitz, A., Bochkareva, E.S. and Girshovich, A.S. (1993). The N terminus of the molecular chaperone GroEL is a crucial structural element for its assembly. J. Biol. Chem. 268, 9957-9959.
Hutchinson, E.G., Tichelaar, W., Hofhaus, G., Weiss, H. and Leonard, K. (1989). Identification and electron microscopic analysis of a chaperonin oligomer from Neurospora crassa mitochondria. EMBO J. 8, 1485-1490.
Ishii, N., Taguchi, H., Sumi, M. and Yoshida, M. (1992). Structure of holochaperonin studied with electron microscopy. FEBS Letters 299, 169-174.
Ishii, N., Taguchi, H., Sasabe, H. and Yoshida, M. (1994). Folding intermediate binds to the bottom of bullet-shaped holo-chaperonin and is readily accessible to antibody. J. Molec. Biol. 236, 691-696.
Jackson, G.S., Staniforth, R.A., Halsall, D.J., Atkinson, T., Holbrook, J.J., Clarke, A.R. and Burston, S.G. (1993). Binding and hydrolysis of nucleotides in the chaperonin catalytic cycle: implications for the mechanism of assisted protein folding. Biochemistry 32, 2554-2563.
Kubota, H., Hynes, G., Carne, A., Ashworth, A. and Willison, K. (1994). Identification of six Tcp-1-related genes encoding divergent subunits of the TCP-1-containing chaperonin. Current Biol. 4, 89-99.
Landry, S.J., Zeilstra-Ryalls, J., Fayet, O., Georgopoulos, C. and Gierasch, L.M. (1993). Characterization of a functionally important mobile domain of GroES. Nature 364, 255-258.
Langer, T., Pfeifer, G., Martin, J., Baumeister, W. and Hartl, F.U. (1992). Chaperonin-mediated protein folding: GroES binds to one end of the GroEL cylinder, which accommodates the protein substrate within its central cavity. EMBO J. 11, 4757-4765.
Marco, S., Carascossa, J.L. and Valpuesta, J.M. (1994a). Reversible interaction of J-actin along the channel of TCP1 cytoplasmic chaperonin. Biophys. J. 67, 364-368.
Marco, S., Ureña, D., Carrascosa, J.L., Waldmann, T., Peters, J., Hegerl, R., Pfeifer, G., Sack-Kongehl, H. & Baumeister, W. (1994b). The molecular chaperone TF55. Assesment of symmetry. FEBS Letters 341, 152-155.
Martin, J., Langer, T., Boteva, R., Schramel, A., Horwich, A. and Hartl, F.-U. (1991). Chaperonin-mediated protein folding at the surface of groEL through a 'molten globule'-like intermediate. Nature 352, 36-42.
Martin, J., Mayhew, M., Langer, T. and Hartl, F.U. (1993). The reaction cycle of GroEL and GroES in chaperonin-assisted protein folding. Nature 366, 228-233.
Phipps, B.M., Hoffman, A., Stetter, K.O. and Baumeister, W. (1991). A novel ATPase complex selectively accumulated upon heat shock is a major cellular component of thermophilic archaebacteria. EMBO J. 10, 1711-1722.
Pushkin, A.V., Tsuprun, V.L., Solojeva, N.A., Shubin, V.V., Evstigneeva, Z.G. and Kretovich, W.L. (1982). High molecular weight pea leaf protein similar to the groE protein of Escherichia coli. Biochim. Biophys. Acta 704, 379-384.
Saibil, H.R. (1994). How chaperonins tell wrong from right. Nature Structural Biology 1, 838-842.
Saibil, H.R., Zheng, D., Roseman, A.M., Hunter, A.S., Watson, G.M.F., Chen, S., auf der Mauer, A., O'Hara, B.P., Wood, S.P., Mann, N.H., Barnett, L.K. and Ellis, R.J. (1993). ATP induces large quaternary rearrangements in a cage-like chaperonin structure. Current Biol. 3, 265-273.
Schmidt, M., Buchner, J., Todd, M..J, Lorimer, G.H. and Viitanen, P.V. (1994). On the role of GroES in the chaperonin-assisted folding reaction. J. Biol. Chem. 267, 10304-10311.
Schmidt, M., Rutkat, K., Rachel, R., Pfeiffer, G., Jaenicke, R., Viitanen, P., Lorimer, G. and Buchner, J. (1994). Symmetric complexes of GroE chaperonins as part of the functional cycle. Science 265, 656-659.
Staniforth, R.A., Burston, S.G., Atkinson, T., and Clarke, A.R. (1994). Affinity of chaperonin-60 for a protein substrate and its modulation by nucleotides and chaperonin-10. Biochem. J. 300, 651-658.
Taguchi, H., Makino, Y. and Yoshida, M. (1994) Monomeric chaperonin-60 and its 50-kDa fragment possess the ability to interact with non-native proteins, to suppress aggregation and to promote protein folding. J. Biol. Chem. 269, 8529-8534.
Todd, M.J., Viitanen, P.V. and Lorimer, G.H. (1994). Dynamics of the chaperonin ATPase cycle: Implications for facilitated protein folding. Science 265, 659-666.
van der Vies, S.M., Gatenby, A.A. and Georgopoulos, C. (1994). Bacteriophage T4 encodes a co-chaperonin that can substitute for Escherichia coli GroES in protein folding. Nature 368, 654-656. [not currently cited]
Weissman, J.S., Kashi, Y., Fenton, W.A. and Horwich, A.L. (1994). GroEL-mediated protein folding proceeds by multiple rounds of binding and release of nonnative forms. Cell 78, 693-702.
Yifrach, O. and Horovitz, A. (1994). Two lines of allosteric communication in the oligomeric chaperonin GroEL are revealed by the single mutation Arg196®Ala. J. Mol. Biol. 243, 397-401.
Zahn, R., Spitzfaden, C., Ottiger, M., Wüthrich, K. and Plückthun, A. (1994). Destabilization of the complete protein secondary structure on binding to the chaperone GroEL. Nature 368, 261-265.
List of Figures
1. line drawing of subunits in oligomer
2. Cryo-EM averaged end and side views of GroEL (a,b) from Chen et. al ., (1994)
3. 3D recontructions of EL, EL ATP, EL ES ATP
4. Colour: backbone & full chain rainbow colour-coded structures of GroEL subunit
5. Ribbon diagrams of GroES structure, top & side views
6. EL-MDH vs EL-ES-MDH subunit domain & hinge diagram
7. Cryo EM of ELES, football side views; EL ES vs EL ES MDH sections
8. Cryo EM section of GroEL vs GroEL-ATP
Figure Legends
1. The cpn60 14-mer. Subunit outlines based on the X-ray crystal structure of GroEL. Diagram adapted from Braig et al. (1994).
2. Cryo EM averaged end and side views of GroEL (a,b), from Chen et al., (1994); and the same views from preliminary data on CCT (c,d) (Chen, Liou, Roseman, Willison & Saibil, unpublished).
3. 3D reconstructions of GroEL, GroEL-ATP and GroEL-GroES-ATP from cryo EM. From work of Chen et al. (1994). (a) Surface-rendered view of the GroEL-GroES-ATP complex at 30 Å resolution determined from cryo EM. The GroES ring is seen as a disk above the GroEL.
4. Atomic resolution structure of the GroEL subunit (Braig et al., 1994). (a) I-carbon and (b) full structure, colour coded in rainbow colours from N (red) to C (blue). In this view, the equatorial domain is the large mass containing both termini at the bottom of the subunit, connected by the antiparallel intermediate domain (cyan and yellow) to the large apical domain (green and cyan) at the top. Images kindly provided by Dr Kerstin Braig.
5. Ribbon diagrams of the GroES structure viewed (a) from above and (b) from the side, showing the J-barrel structure of the subunit with a J-hairpin forming the roof of the dome-like heptamer. The N and C termini are labelled and and point radially outwards. In (a), the positions of two glutamic acid residues in the J hairpin are indicated as negative charges. The broken ends of the disordered mobile loop, which is not seen in the map, are indicated by dashed arrows in (b). Images kindly provided by Dr John Hunt.
6. Schematic diagram of the subunit arrangement in a hypothetical slice through the oligomer, showing the major functional sites. (a) GroEL, based on the crystal structure with bound substrate (shaded light to dark) as imaged by cryo EM. A, apical domain; I, intermediate domain; E, equatorial domain. The apical domains form a ring of hand-like structures with the substrate binding sites on the fingers protruding into the central channel. Sites of potential hinge rotations are indicated by black dots. The notch in the equatorial domain represents the ATP-binding cleft. The interaction across the equatorial plane is shown as pairs of wavy surfaces. (b) the folding complex GroEL-MDH-GroES-ATP, rotating the subunit domains and adding the GroES (dark gray) and substrate (shaded) densities according to cryo EM observations.
7. (a) Side view of the GroEL-GroES complex averaged from 200 cryo EM images (Chen et al, 1994). (b) Side view average of "football" complexes from cryo EM (Chen, S., Roseman, A., Saibil, H., unpublished observations). (c) Section through the complex in (a) showing the large reorientation of the apical domains in contact with GroES, forming the contact with the same region as the substrate binding site. The GroEL cylinder is greatly elongated. (d) section of a 3D reconstruction of the folding complex GroEL-MDH-GroES-ATP trapped by vitrification after 12 sec of folding from cryo EM (Chen et al., 1994). The MDH substrate density is found in the opposite ring to GroES.
8. Superposed outlines of sections through GroEL and GroEL-ATP (shaded) from cryo EM (Chen et al, 1994), showing the slight but distinct outward rotation of the apical domains in the ATP form. These displacements have the effect of lengthening the cylinder and widening the binding cavity.