INTRODUCTION
The determination of the forces that govern protein
stability is of fundamental importance for our tability to understand and control the interactions of
complex biological molecules. It has been known since the 1960s that the primary structure of a protein dictates its three dimensional fold, yet a comprehensive understanding of the factors that impart thermodynamic stability to proteins is elusive. This is because the tertiary folds of native proteins are defined by a large number of weak interactions: hydrogen bonding, hydrophobic interactions, salt bridges, and weakly polar interactions. In addition to these noncovalent forces, proteins are also stabilized covalently by disulfide bonds.
The overwhelming majority of data concerning the stability of proteins comes from examinations of the
reversible denaturation of small, globular proteins.
The stability of both the wild type and mutant proteins is expressed as the melting temperature, Tm,
which is obtained by studying reversible heat denaturation.
For many monomeric, single-domain denaturation by heat or chemicals is often consistent with a single, cooperative transition from the folded, N-state to a mostly disordered form, the D-state. This two-state model can be written as:
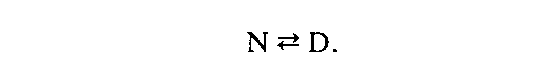
The difference in free energy between the D and N states is defined as Gd, which is equivalent to –RT ln [D]/[N] . Therefore, the more stable the protein, the more positive will be. The advent of molecular biology and the ability to generate varient proteins without excessive difficulty has spawned an intense reinvestigation of the factors that affect this free energy.