
Instruction: In this document you can catch the advantages and disadvantages of the two mentioned methods with different colours of highlighting (the NMR is bolded and the X-ray is in italics). At the end of the document, in the Summary section you will get a collection about these features.
Contents:
3. An overview of the X-ray crystallography
1. Introduction
There are several methods by which one can get information about the proteins. Different methods are related to different types of information. We can think to e.g. the gel filtration or Western blotting where we obtain information from the macroscopic level of the protein (size, molecular mass, binding to an antibody etc.). One may find information about the proteins with several chemical modifications (or even mutations in the DNA - e.g. site-directed mutagenesis) and checking what kind of change occurred one could draw a conclusion for the structure (e.g.: active center prediction - with the investigation of substrate-specificity - , side-chain properties etc.). These experiments lead us closer to the real structure, but we could not be informed enough for being sure that different subunits how are arranged into a 3D construction. And we do not know from this level of examinations lots of different sorts of information needed to predict the whole structure and the way of the mechanism by which it is working (e.g. as an enzyme).
On this deeper level we need methods refered to the smallest
parts of the structure. This level is the atomic or even the electron
level. Albert Szent-Györgyi said that the biologists would
understand the mechanism of life if they realized the importance
of the electronic level of Biology. Now, we have some suitable methods
which are capable to resolve these levels. We can mention first of all
the different spectroscopies which are related to different quantated energy
levels. The problem of handling these different energy levels seems to
be appeared in all of the spectroscopic examinations but from the energy-difference
of these different energy-levels we are able to say which kind of transitions
occured and which band is devoting to this changing in that spectrum. Almost
every transition is known in every spectroscopic method, so one can conclude
the existence or nonexistence of a given property or functional group in
that protein (or in any other material). If we go through the electromagnetic
spectrum we could find several ranges whose applications are capable to
tell us the existence of these different features. Thus, we might take
into account lots of methods e.g.: Rotation-, Infrared-, Raman-, Ultraviolet-,
CD-, Electron- etc. spectroscopies.
2. An overview of the NMR
In the electromagnetic spectrum there is a range which
came to light just about 40 years ago. This range is the radiowave region.
The energy of this wave is able to excite the spin-moments of the nucleons
in the nucleus or that of the electrons on the shell depending on its wavelength.
Let us see the nucleus.
Since 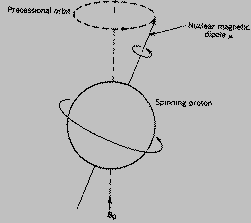 the
nucleus is surrounded by electrons that's why this energy - the strength
of the electromagnetic field (pulse) - needed for excitation is changing
depending on (first of all) the structure of the electron shell of that
atom. If the atoms are in connection with different atoms in a chemically
bonded molecule by their outer valence electrons this new electron distribution
also changes the electromagnetic field around the nucleus. In this way
the transition energy may change again since it is the function of the
strength of the electromagnetic field around the given nucleus. That's
why if we go through a range of wavelengths around the average transition
energy we can get the absorbtions as a function of the wavelength. This
is the NMR spectrum. From the extent of the shift and the efficiency of
transmission we can conclude which type of and how many atoms or particles
are connected directly to that atom or in which type of environment this
is. This way the chemical surrounding or environment can be solved.
Since the only one factor which makes difference in the spectrum is the
difference in the strength of the electromagnetic field around the nucleus
that's why in this method (the name of which is NMR - Nuclear Magnetic
Resonance) there could be (and are) several methods which play with this
principle to get several types of information
from lots of types of experiments (with programmed impulses
of the magnetic field). In this way they obtain
angles, distances, coupling constants, chemical shifts, rate constants
etc. These are really molecular parameters which could be examined more
with computers and molecular modeling procedures. Since these
parameters are related to the atoms and their surroundings, if
we have enough strength of the magnetic field (the resolution is the function
of that) than we can handle all of the atoms personally. With
a suitable computer apparatus we can calculate the whole 3D structure.
But we have lots
of atoms and a lot of extracted data from a system. This is good for the
more accurate determination of the structure, but not for the availability
of higher molecular masses. That's why,
the resolving power of NMR is less than some other type of experiments
(e.g.: X-ray crystallography) since the information got from the same material
is much more complex. The cost of the
experimental implementation is increasing with the higher strength and
the complexity of the determination. This is because the NMR
hours are very expensive since the technical apparatus needs supporting
conditions which make the cost high (e.g. maintaining the very low temperature
in the magnetic part) . This problem can be solved by the progress in the
technology (e.g.: increasing the available strength of the field). Nowadays
the highest molecular mass which was examined successfully is just a
64kDa protein-complex. There is another problem which is connected
to these thinkings. There are lots of possibilities
to collect different data-sets from different types of experiments for
the ability to resolve the uncertainties of one type of measurements because
there are lots of cases when from a given
data-set - a given type of experiment - we could predict two or more possible
conformations, too. In these cases, unfortunately, we are just able to
determine the degree of probability of being of the protein segment in
the given conformation. That`s why the variability of the achievement
of the principles in different methods is very useful.
the
nucleus is surrounded by electrons that's why this energy - the strength
of the electromagnetic field (pulse) - needed for excitation is changing
depending on (first of all) the structure of the electron shell of that
atom. If the atoms are in connection with different atoms in a chemically
bonded molecule by their outer valence electrons this new electron distribution
also changes the electromagnetic field around the nucleus. In this way
the transition energy may change again since it is the function of the
strength of the electromagnetic field around the given nucleus. That's
why if we go through a range of wavelengths around the average transition
energy we can get the absorbtions as a function of the wavelength. This
is the NMR spectrum. From the extent of the shift and the efficiency of
transmission we can conclude which type of and how many atoms or particles
are connected directly to that atom or in which type of environment this
is. This way the chemical surrounding or environment can be solved.
Since the only one factor which makes difference in the spectrum is the
difference in the strength of the electromagnetic field around the nucleus
that's why in this method (the name of which is NMR - Nuclear Magnetic
Resonance) there could be (and are) several methods which play with this
principle to get several types of information
from lots of types of experiments (with programmed impulses
of the magnetic field). In this way they obtain
angles, distances, coupling constants, chemical shifts, rate constants
etc. These are really molecular parameters which could be examined more
with computers and molecular modeling procedures. Since these
parameters are related to the atoms and their surroundings, if
we have enough strength of the magnetic field (the resolution is the function
of that) than we can handle all of the atoms personally. With
a suitable computer apparatus we can calculate the whole 3D structure.
But we have lots
of atoms and a lot of extracted data from a system. This is good for the
more accurate determination of the structure, but not for the availability
of higher molecular masses. That's why,
the resolving power of NMR is less than some other type of experiments
(e.g.: X-ray crystallography) since the information got from the same material
is much more complex. The cost of the
experimental implementation is increasing with the higher strength and
the complexity of the determination. This is because the NMR
hours are very expensive since the technical apparatus needs supporting
conditions which make the cost high (e.g. maintaining the very low temperature
in the magnetic part) . This problem can be solved by the progress in the
technology (e.g.: increasing the available strength of the field). Nowadays
the highest molecular mass which was examined successfully is just a
64kDa protein-complex. There is another problem which is connected
to these thinkings. There are lots of possibilities
to collect different data-sets from different types of experiments for
the ability to resolve the uncertainties of one type of measurements because
there are lots of cases when from a given
data-set - a given type of experiment - we could predict two or more possible
conformations, too. In these cases, unfortunately, we are just able to
determine the degree of probability of being of the protein segment in
the given conformation. That`s why the variability of the achievement
of the principles in different methods is very useful.
(Of course, in the X-ray experiments we have also probability
factors, but only in the meaning of determining an average conformation
which has a probability for its existance, but this is sure that it is
existed in some percentage). The more experiments are made the more precise
determination or conclusion could be drawn. Since
the motion of the segments (domains)
changes the parameters of the atoms in the whole structure, that's why
it can also be
examined by NMR. For this exploration
we need a comfortable impulse for the NMR experiment with which we can
follow the time-dependence of this parameter-set. So, this
method is capable to lead us for the observation of the chemical kinetics.
Since e.g. a chemical reaction is a kind of motion because the ligands
(substrates, reagents) are going forward to another chemical environment
(exchange mechanism) the kinetics of that reaction and all of the (activation-)thermodinamic
(and certainly kinetic) data could be determined from a well-prepared (dynamic-)NMR
experiment. If we realize that the actual conformation depends
on the environment of the protein molecule by the NMR we
can investigate the influence of the dielectric constant, the polarity
and any other properties of the solvent or some added material.
In this context this is one of the best methods which
can solve the 3D structure of a protein.
Here is a tipical 13C NMR spectrum
(but not for a protein, since it would be very confused and complex (so,
it is just for illustration):
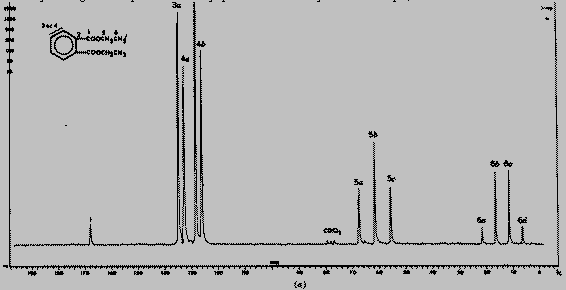
If you want to have a more precise explanation of NMR
principles just click here.
(http://www.ch.ic.ac.uk/local/organic/nmr.html)
And you should see this one: (http://www.cryst.bbk.ac.uk/PPS2/projects/schirra/html/intro.htm)
and also this
one: (http://www.cryst.bbk.ac.uk/PPS2/projects/schirra/html/theory.htm)
3. An overview of the X-ray
crystallography
There is another principle which can lead us to get a
similar type of parameter-set of atoms. This principle is of diffraction.
There are some types: neutron-, electron-and X-ray-diffraction. Since in
these methods the
crystal structure is necessary only the proteins which can be crystallized
are examinable. We can also examine
by this way the solvent effect since from different solvents the same protein
may crystallize into different crystalloid form. Thus, we are able
to force the protein to an other form of crystallization by the change
of its solvent.
Accordingly, this procedure is in solid phase, and that's
why we cannot examine solutions and the behavior
of the molecules in solution. If we try, it we get rings instead
of diffraction spots since these systems have cylindrical symmetry and
these kind of data are not considerable for the determination of chemical
structure. This happens when we try to examine
powders, gases ,also. The  studying
of motions is not available, but they wrote about some trials
in which the average electron distribution would be resolved for studying
the motions in the molecule (ultra fast diffraction - catching atoms
in their flight). But it is only the future of this method.
studying
of motions is not available, but they wrote about some trials
in which the average electron distribution would be resolved for studying
the motions in the molecule (ultra fast diffraction - catching atoms
in their flight). But it is only the future of this method.
There are some methods by which the structure of different
surfaces can be studied.
The principle itself is based on the interference of
two or more different waves. There can be constructive and destructive
waves in this way (in phase and in anti-phase, respectively). After the
irradiation of the sample the scattering waves will interfere and it is
at the detector where we get the constructive and destructive interfered
waves depending on the difference of distances of the two or more atoms
from the detector on their paths. For this reason we would need a wavelength
which is comparable with the atomic distances for the comfortable examination
of difference in distances we should choose the range of hard X-ray, because
its typical wavelength is 10-10 m.
There are other possibilities: the matter waves.
Their suitable wavelengths can be calculated by the De Broglie equation.
There are two possibilities: a., thermal neutrons
, b., electrons.
If we examine crystals they produce sharp spots at the
detector corresponding to their structure. There are several rotation angles
of the crystal in the diffractometer in order to get information from lots
of angles. The set of diffraction points is analyzed to obtain the electron
density of the compound and then the chemical structure is fitted
to that. By this way we could get the whole 3D
structure by the systematic analysis of a good crystallized material.
Since from the X-ray experiment we can get only
one parameter-set, we are able to observe only one conformation
versus NMR as we mentioned above. Because of the basic principles of X-ray
crystallography there is no possibility to examine
small parts in the molecule. There is no chance for direct determination
of secondary structures and especially domain movements (big disadvantage
against the NMR). Because the X-ray diffraction is based on
the connection of the scattered waves with the electron density, the
hydrogen in the molecules are not examinable since it has only one electron
and this has no enough efficiency for scattering and its detection. That's
why we need another method for the detection of hydrogen (e.g.: neutron
diffraction, NMR, ect.).
This is a tipical picture of a structure fitted to the electron
density (just for illlustration):
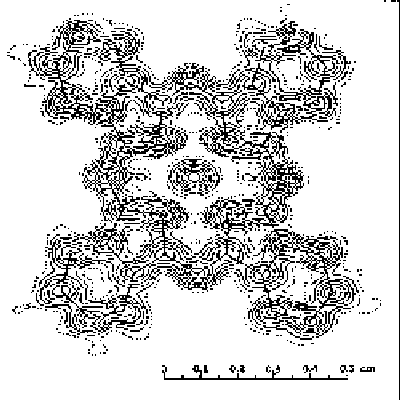
If you want to have a deep explanation on basic theories just click here (http://www-wilson.ucsd.edu/education/xraydiff/xraydiff.html)
or here
(http://wserv1.dl.ac.uk/SRS/PX/rck/2_pps96/project/index.html)
4. What kind of data can
be collected exactly from NMR for the determination of secondary structure
of proteins?
(http://iona.cryst.bbk.ac.uk/course/section8/ss-960531_22.html)
2. A more experimental explanation
(http://www.cryst.bbk.ac.uk/pps2/projects/schirra/html/1dnmr.html)
5. What kind of data can
be collected exactly from X-ray crystallography for the determination of
3D structure of proteins?
Click
here
(http://wserv1.dl.ac.uk/SRS/PX/rck/2_pps96/project/index.html)
6. Summary: The comparison
The collection of advantages and disadvantages (or strengths
and weaknesses) of NMR and X-ray diffraction.
| Advantages of NMR | Disadvantages of NMR | Advantages of X-ray | Disadvantages of X-ray |
| 1. several types of information from lots of types of experiments | 1. we have lots of atoms and a lot of extracted data from a system. | 1. . We can examine also by
this way the solvent effect since from different solvents the same protein
may crystallize into different crystalloid form.
|
1. the crystal structure is necessary only that proteins which can be crystallized are examinable |
| 2. they obtain angles, distances, coupling constants, chemical shifts, rate constants etc. These are really molecular parameters which could be examined more with computers and molecular modeling procedures. | 2. This is good for the more accurate determination of the structure, but not for the availability of higher molecular masses | 2. So we are able to force the protein to an other form of crystallization by the change of its solvent. | 2. we cannot examine solutions and the behavior of the molecules in solution |
| 3. if we have enough strength of the magnetic field (the resolution is the function of that) than we can handle all of the atoms personally | 3. the resolving power of NMR is less than some other type of experiments (e.g.: X-ray crystallography) since the information got from the same material is much more complex | 3.we could get the whole 3D structure by the systematic analysis of a good crystallized material | 3. This happens when we try to examine powders, gases |
| 4. With a suitable computer apparatus we can calculate the whole 3D structure | 4. the highest molecular mass which was examined successfully is just a 64kDa protein-complex | 4. studying of motions are not available | |
| 5. There are lots of possibilities to collect different data-sets from different types of experiments for the ability to resolve the uncertanities of one type of measurements | 5. there are lots of cases when from a given data-set - a given type of experiment - we could predict two or more possible conformations, too | 5. we can get only one parameter-set so we are able to observe only one conformation | |
| 6. the motion of the segments (domains) can be examined | 6. unfortunately we are just able to determine the degree of probability of being of the protein segment in the given conformation | 6. there is no possibility to examine small parts in the molecule | |
| 7. this method is capable to lead us for the observation of the chemical kinetics | 7. The cost of the experimental implementation is increasing with the higher strength and the complexity of the determination | 7. There is no chance for direct determination of secondary structures and especially domain movements (big disadvantage against the NMR) | |
| 8.(activation-)thermodinamic (and certainly kinetic) data could be determined from a well-prepared (dynamic-)NMR experiment | 8. the hydrogen in the molecules are not examinable since it has only one electron | ||
| 9. we can investigate the influence of the dielectric constant, the polarity and any other properties of the solvent or some added material |
7. Conclusion
As a conclusion we must say that the two methods are very
basic and critical in the field of protein structure determination. The
strengths and weaknesses of one of the two methods fortunately are of those
kinds which supplement the holes and gaps in the other method to make it
possible that different kind of important data for a structural question
can be answered with the parallel or supplemental application of the two
methods. There may be several instances when only one method could be used.
If the two methodologies had the same basics, principles, strengths and
weaknesses we may be not capable to solve lots of structural problems.
We should thank this possibility to the technical development which could
catch different biophysical principles.
8. References
3. Structure
determination of proteins with NMR spectroscopy
by Horst Joachim Schirra
(http://www.cryst.bbk.ac.uk/pps2/projects/schirra/html/home.htm)
4. Magnetic
Resonance
by László
Szilágyi (in hungarian), 1987,
University of
Lajos Kossuth (KLTE), Hungary
5.
The investigation of ligand exchange processes of cyanide ion
6. X-ray Diffraction
7. Diffraction
methods (in hungarian)
8. And some
valuable conversations with an expert molecular modellist:
9. Acknowledgments
to:
in the HCN-DCN and the Tl(edta)CN2- - CN- systems
by
dynamic NMR spectroscopy (in hungarian)
by Attila Ambrus,
Diploma Thesis, 1996, supervisor: István
Bányai,
Dept. of Physical Chemistry, University
of Lajos Kossuth, Hungary
http://www-wilson.ucsd.edu/education/xraydiff/xraydiff.html)
by
Ferenc Joó,
Dept.
of Physical Chemistry, University of Lajos Kossuth, Hungary
Péter Bagossi