4.3 Prediction of Secondary Structure
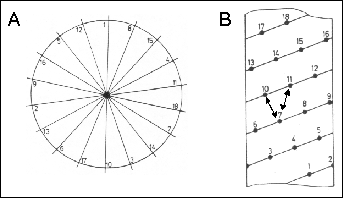
Figure 34. Helical wheel (A) and helical net (B) representations for locating amphiphilic helices and other intra-helical interactions. Arrows in (B) indicate the (i, i+3) and the (i, i+4) sidechain interactions enforced by the helical conformation.
Many different algorithms have been proposed for the identification of amphiphilic segments. Finer-Moore & Stroud (1984) used a Fourier analysis and Eisenberg et al. (1982) used a vector analysis of hydrophobicities to locate these amphiphilic structures. Although amphiphilicity is a strong organizing force (3.1.4.2), not all helices or sheets contain this motif and not all polypeptide segments with amphiphilic potential (i.e., helix or sheet) are not in the conformation expected from the amphiphilic analysis.
The algorithm of Kyte & Doolittle (1982) has been particularly successful in locating membrane-spanning segments and exposed versus buried portions of soluble globular proteins. The method involves plotting the amino acid hydrophobicity as a function of the amino acid sequence of the protein in question. Polypeptide segments spanning membranes or in the interior of globular proteins appear as continuous segments of hydrophobic residues whereas loops and turns appear as continuous segments of hydrophilic residues. This algorithm has been extensively used in identifying potential immunogenic sequences in proteins.
Amino acid hydrophobicity can be defined in many ways. Based on empirical and theoretical considerations dozens of hydrophobicity scales have been reported the literature (e.g., see Cornette et al., 1987 for a compilation of 46 such scales). The results obtained from these stereochemical based prediction methods will depend to a large extent on the particular scale used. These differences in these hydrophobicity scales are primarily in the ordering of hydrophobic residues. A prime example of discrepancies in the various scales is that Trp, Tyr and Phe may be classified as the most apolar (e.g., Levitt, 1976) or even among the polar residues (Wolfenden et al., 1981).
Generated with CERN WebMaker
No Title - 31 MAY 96
written by Kurt D. Berndt