Physical principles of NMR
Sub-atomic particles (e.g., proton, neutron, electron, etc.) possess a characteristic called spin angular momentum. From quantum mechanics, each particle has a spin value of 1/2. The combination of multiple particles in the nucleus results in an overall spin property for each atomic isotope. Those isotopes with an even number of protons and neutrons will have zero magnetic spin (e.g., He-4, C-12 and O-16). An odd number of protons and an even number of neutrons (e.g., H-1, N-15, or F-19) or an odd number of neutrons and an even number of protons (e.g., He-3, O-17 or Ca-41) result in an overall (multiple of 1/2) spin. Those isotopes with odd numbers of both protons and neutrons (e.g., H-2 or N-14) have more complex spin states and are less suitable for direct NMR observation in macromolecules.Fortunately, each of the four most abundant elements in biological material (H, C, N, and O) have at least one naturally occurring isotope with non-zero nuclear spin and is in principle observable in an NMR experiment. The naturally occurring isotope of hydrogen, H-1, is present at >99% abundance and forms the basis of the experiments described here. Other important NMR-active isotopes include C-13 and N-15 present at 1.1 and 0.4% natural abundance, respectively. The low natural abundance of these two isotopes makes their observation difficult on commonly isolated natural products. These two nuclei are however very extensively used for larger (>10 kD) proteins which can be isotopically enriched (to >95% if necessary) when cloned into over expression systems.
In the presence of an external magnetic field, the spin angular momentum of nuclei with isotopes of overall non-zero spin will undergo a cone-shaped rotation motion called precession. The rate (frequency) of precession for each isotope is dependent on the strength of the external field and is unique for each isotope. For example, in a magnetic field of a given strength (e.g. 14.1 Tesla) all protons in a molecule will have characteristic resonance frequencies (chemical shifts) within a dozen or so parts per million (ppm) of a constant value (e.g., 600.13 MHz) characteristic of the particular nuclear type. These slight differences are due to the type of atom the proton is bound (e.g., C, N, O, or S) and the local chemical environment. Thus each proton should, in principle, be characterized by a unique chemical shift. In practice, this is never observed as some protons such as the three protons of each sidechain methyl group of Thr, Val, Leu, Ile, and Met and most pairs of equivalent (2,6 and 3,5) aromatic ring protons are found to have degenerate chemical shifts. Other protons (e.g., some OH, SH, and NH3) are in rapid chemical exchange with the solvent and thus have chemical shifts indistinguishable from the solvent resonance. Nevertheless, nearly complete chemical shift assignments are often possible and are a prerequisite for structural studies by NMR.
Structural information from NMR experiments come primarily from through-bond (scalar or J coupling) or through space (the nuclear Overhauser effect NOE) magnetization transfer between pairs of protons. J couplings between pairs of protons separated by three or fewer covalent bonds can be measured. The value of a three-bond J coupling constant contains information about the intervening torsion angle. This is called the Karplus relationship and has the form:
3J = A cos (theta) +B cos2 (theta) + C
where A, B, and C are empirically derived constants for each type of coupling constant (e.g., 3JHAHN or 3JHAHB). Unfortunately in general, torsion angles cannot be unambiguously determined from a Karplus-type relationship since as many as four different torsion angle values correlate with a single coupling constant value as seen below in Figure 27 .Similar relationships can be determined between the three-bond coupling constant between the alpha proton and the beta proton(s) yielding information on the value of the sidechain dihedral angle chi1. Constraints on the dihedral angles phi and chi1 are important structural parameters in the determination of protein three-dimensional structures by NMR.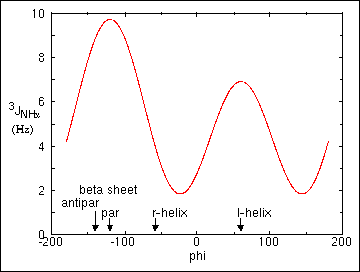
Figure 27. The empirically-derived Karplus relationship between the vicinal three-bond coupling constant 3JHNa and the intervening torsional angle phi.
The other major source of structural information comes from through space dipole-dipole coupling between two protons called the NOE. The intensity of a NOE is proportional to the inverse of the sixth power of the distance separating the two protons and is usually observed if two protons are separated by < 5 Angstroms. Thus the NOE is a sensitive probe of short intramolecular distances. NOEs are categorized according to the location of the two protons involved in the interaction. Intraresidual NOEs are between protons within the same residue whereas sequential, medium, and long range NOEs are between protons on residues sequentially adjacent, separated by 1, 2 or 3 residues, and separated by four or more residues in the polypeptide sequence. A network of these short inter-proton distances form the backbone of three-dimensional structure determination by NMR.
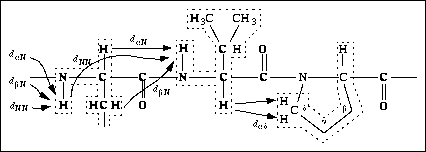
Figure 28. Segment of a polypeptide chain with indication of the sequential NOE connectivities with amino acids and with proline (shown in the trans conformation). Stippled lines outline independent amino acid spin systems.
right-handed alpha helix, phi = -57, 3JHAHN = 3.9 Hz
right handed 3.10 helix, phi = -60, 3JHAHN = 4.2 Hz
antiparallel beta sheet, phi = -139, 3JHAHN = 8.9 Hz
parallel beta sheet, phi = -119, 3JHAHN = 9.7 Hz
left-handed alpha helix, phi = 57, 3JHAHN = 6.9 Hz
Figure 29. Sequential distance between the alpha proton of residue i and the amide proton of residue i+1 as a function of the backbone dihedral angle psi. The solid curve is drawn for a L-amino acid in position i and the broken curve is drawn for Gly in position i. The solid black bar at the bottom indicates the allowed psi values for non-Gly amino acids. The positions of the right-handed alpha helix and the parallel and the antiparallel strands are also indicated. This figure was taken from Wüthrich, 1986.
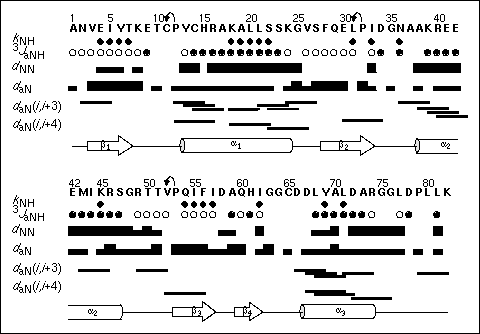
Figure 30. Survey of NMR-derived structural parameters characterizing reduced Grx3 in solution at pH 6.8 and 28 xfb C as a function of the amino acid sequence. The following features are identified: amide proton exchange rates with solvent water (filled diamonds) kNH < 0.02 min-1; coupling constants 3JHNa (filled circles) < 6.0 Hz, (open circles) > 7.0 Hz; sequential backbone dNN and daN NOE connectivities are classified as strong, weak, or absent and are represented by the thickness (or absence) of a bar connecting the residues in question; medium range NOE connectivities daN (i, i+3) and (i, i+4) are drawn as line segments connecting the residues contributing to the observed crosspeak if present. Arrows above the sequence connecting a proline to the previous residue indicates a sequential connectivity via either sequential daa or sequential dad NOEs. All NOEs were measured in a 2D NOESY spectrum recorded with 60 ms mixing time. The figure is taken from Åslund et al., 1996.
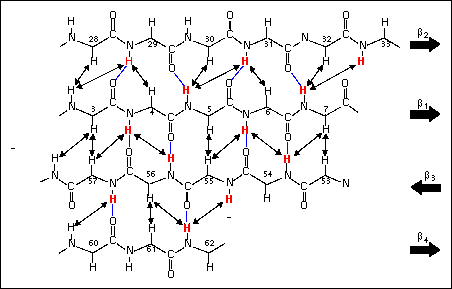
Figure 31. Diagram showing the alignment of the four-stranded b sheet observed in Grx3 deduced from interstrand NOE connectivities. The amino acid backbone is represented in stick format with residue numbers indicated above Ca positions. Interstrand NOEs are drawn as double-headed arrows connecting protons giving rise to the observed crosspeaks. Slowly exchanging (kNH < 0.02 min-1 ) amide protons are identified in red. Hydrogen bonds suggested by the network of NOEs are drawn as solid blue lines connecting amide proton and acceptor carbonyl oxygen. The directionality of individual strands of the b sheet is indicated by bold arrors at the right of the figure. The figure is taken from Åslund et al., 1996.
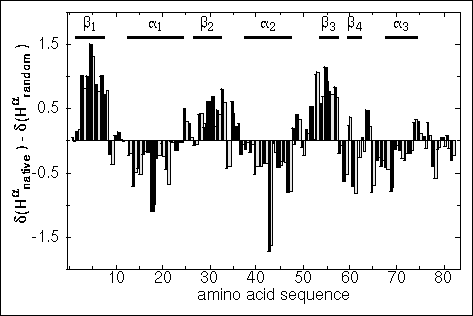
Figure 32. Plot of the differences between the observed alpha proton chemical shifts and the corresponding random coil values, d(Hanative) - d(Harandom), versus the amino acid sequence of Glutaredoxin 3 (Åslund et al., 1996). Two vertical bars representing differences calculated using (i) average random coil alpha proton chemical shifts taken from Wishart et al. (1992) (black), and (ii) experimentally determined random coil alpha proton chemical shifts for reduced Grx3 taken from Nordstrand et al. (1995) (stippled), are centered on the amino acid residue position. Regions of secondary structure identified by analysis of NOEs, J-couplings and amide proton exchange rates are indicated by solid black line segments at the top of the plot.
Figure 33. Survey of the sequential and medium range proton-proton NOE's and the spin-spin coupling constants 3JHNa in some common secondary structures. The numbers at the bottom represent the amino acid residues in the given secondary structure and the values of the 3JHNa coupling constant. Short proton-proton distances are indicated by lines linking the residues that contain the hydrogen atoms involved. The thickness of the line is proportional to the intensity of the NOE. This figure was taken from Wüthrich, 1986.
Generated with CERN WebMaker
Secondary structure from NMR parameters
Secondary structure determination by NMR techniques does not require a full three-dimensional structural analyses as does X-ray crystallography. Knowledge of the amide and alpha proton chemical shifts are in principle all that is necessary although if this information is available it is likely that nearly complete assignments of sidechain protons are also available. While obtaining the sequential resonance assignments is a laborious task, the NMR method is perhaps the most powerful and certainly the most accurate method of secondary structure determination without a three-dimensional structure. Coupling constants
The three-bond coupling constant between the intraresidual alpha and amide protons is the most useful for secondary structure determinations as it can be directly related to the backbone dihedral angle phi. Unfortunately, no there is no three-bond proton coupling that can be related to the angle psi. As seen in the previous sections, helical and extended conformations have very different values for phi (-60 and -120, respectively) which result in differences in 3JHAHN. NOEs
A number of short (< 5 Angstroms) distances are fairly unique to secondary structural elements. For example, alpha helices are characterized by short distances between certain protons on sequentially neighboring residues (e.g., between backbone amide protons, dNN, as well as between beta protons of residue i and the amide protons of residue i+1, dBN. Helical conformations result in short distances between the alpha proton of residue i and the amide proton of residues i+3 and to a lesser extent i+4 and i+2. These i+2, i+3, and i+4 NOEs are collectively referred to as medium range NOEs while NOEs connecting residues separated by more than 5 residues are referred to as long range. Extended conformations (e.g., beta strands) on the other hand, are characterized by short sequential, dAN, distances. The formation of sheets also result in short distances between protons on adjacent strands (e.g., dAA and dAN).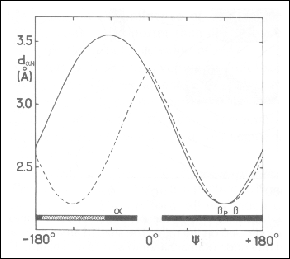
Amide proton exchange rates
The regular hydrogen-bonded secondary structures "protect" amide protons involved in them as evidenced by their significantly reduced amide proton exchange rates with the solvent (H2O). Although nearly all polypeptide amide protons are involved in hydrogen bonds in a globular protein (Baker & Hubbard, 1984) those in regular secondary structures appear to be longer-lived. For example, after placing a lyophalized sample of BPTI into 2H2O many amide protons are completely replaced with deuterons within 1hr. Over the next several hours, the amide protons in the N-terminal and then the C-terminal helix also completely exchange. However, some amide protons participating in the central antiparallel sheet are still present after some months. Selection of secondary structural segments
Unfortunately, no single criterion (coupling constants, slowed amide proton exchange, or short and medium-range NOEs) is sufficient for an unambiguous assignment of secondary structure. For example, short distances (< 3.6 Angstroms) between sequentially neighboring amide protons (dNN) are a necessary consequence of a helical conformation but the presence of a short sequential dNN is not a sufficient criterion for helical prediction as 49% of such occurrences are not in helical conformations (Wüthrich, 1986). However, sequential stretches of residues with consistent secondary structure characteristics (NOEs, coupling constants, slowly exchanging amide protons, and chemical shifts) provide a reliable indication of the location of these structural segments. However, the boundaries of these segments are difficult to define precisely. 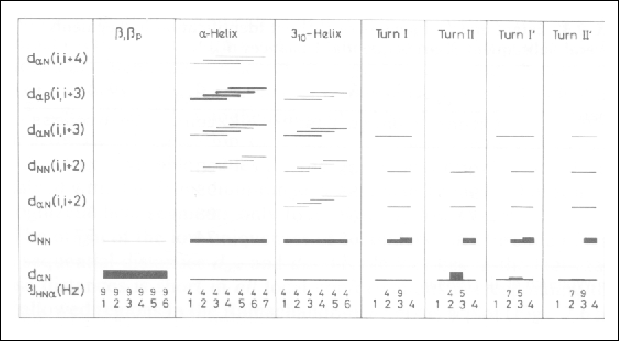
No Title - 31 MAY 96
written by Kurt D. Berndt