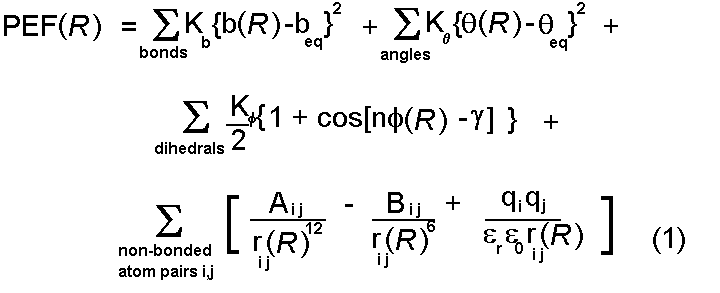
We have briefly reviewed the variety of interactions which
are important in protein interactions and seen suitable simple mathematical
forms for their representation. These are drawn together to form a potential
energy function:
This function can be used to calculate a value for the potential energy
(PEF(R)) for any conformation of a given protein - defined by the
(normally Cartesian) coordinate vector R. A number of important points
can be made:
This section provides a very brief introduction into what the uses of potential energy functions are in protein studies.
This is in many ways the simplest simulation procedure. The basic idea is that starting from some structure (R we find its potential energy using the potential energy function given as equation (1) above. The coordinate vector R is then varied using an optimization procedure so as to minimize the potential energy PEF(R).
Very often these methods are used if a distorted structure is produced - e.g. a homology based model. Energy minimization can then relieve short interatomic distances while maintaining important structural features.
Energy minimization can be used to help to solve experimental structures:
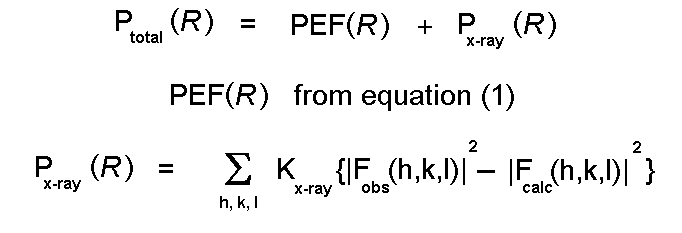
The program XPLOR is commonly used to do this.
Extensive notes on optimization procedures are available from the M.Sc. Molecular Modelling and Bioinformatics at Birkbeck.
In molecular dynamics studies the motion of a molecule is simulated as a
function of time. A simple description is that Newton's second law of
motion:
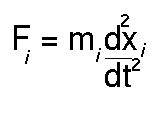
is solved to find how the position for each atom of the system xi varies with t. To find the forces on each atom (Fi) the derivative vector (or gradient) of equation 1 is calculated. Factors such as the temperature and pressure of the system can be included in the treatment.
Molecular dynamics simulation procedures are very popular in the protein field. They have the advantage that they can treat systems where motion is essentially diffusive in character - important because of the role of water in protein structure. The procedures can be used to calculate "ensemble average" properties - recent advances have included the ability to calculate free energy differences between (slightly) different ligands or conformations of a protein. A disadvantage of conventional molecular dynamics procedures is that they can only tackle motions with a relatively short time scale - one nanosecond is the approximate upper limit with current computers.
Very many other methods use potential energy functions for the study of proteins conformation and dynamics. An example of this is the Path Energy Minimization procedure - which aims to find routes for large scale conformational transitions of proteins (developed by the author of this section of the course).
Useful sources of information to start to learn more about this topic:
© O.S. Smart 1996, all rights reserved
Back to main Molecular Forces index
Back to previous unit The effect of solvent and hydrophobic interactions