This project was made for PPS'97 examination
Jacek Leluk
Institute of Biochemistry and Molecular Biology
University of Wroclaw
Wroclaw
Poland
e-mail: lulu@bf.uni.wroc.pl
SOME BASIC INFORMATION ABOUT CYSTEINE ITSELF
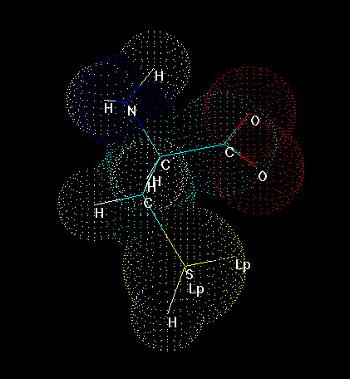
Cysteine (Fig. 1) is one of 20 naturally occurring, 'biogenic' amino acids which linked by peptide bonds form polypeptides and proteins. Like the other amino acids cysteine is abundant as L-form. It is genetically encoded by two possible codons (nucleotide triplets of mRNA) UGU and UGC. Structurally cysteine belongs to the sulfur amino acids, because of sulfur atom appearing in its side chain. The sulfur atom of cysteine is involved in formation the sulfhydryl group which is very reactive. That differs cysteine from another sulfur amino acid - methionine which has a methyl group attached to the sulfur. Thus methionine is more hydrophobic, sterically larger and much less reactive than cysteine. Cysteine can be easily oxidized to form a dimer containing disulfide bridge between two cysteines. Such dimer is known as cystine (Fig. 2) and this feature is very important for analysis the primary structure of proteins, for effects on changes in secondary structure and for stabilization tertiary and quaternary structure. Sulfhydryl group of cysteine can be considered also as a very strong reducing factor, which is very important for activity of many proteins, a oligopeptides (glutathione), has a strong influence on redox potential of environment in certain compartments in vivo, and can form a center of the active site of some enzymes (e.g. thiol proteinases).
Fig.1. Cysteine
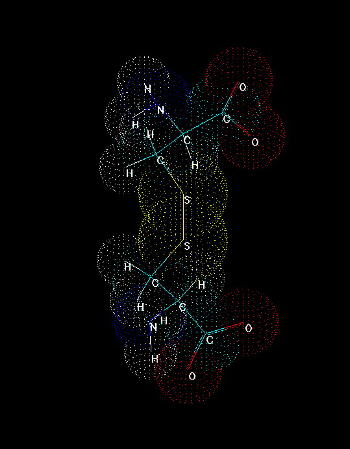
Fig.2. Cystine. A dimer of two cysteines linked by disulfide bridge.
The presence of sulfhydryl group where hydrogen can be easily replaced by radicals and other groups, makes it possible to form a covalent bond with the other molecules. Cystine formation is an example of such activity. If a sulfur atom is bonded to sulfur atom of another cysteine, a covalent disulfide bridge is formed which is weaker (easier to split) than peptide bond, but stronger than any interaction of the other type (hydrogen bonds, salt bridges, hydrophobic and van der Waals interactions). The easiness of formation of such covalent bond depends on overall redox potential of environment as well as pH (at low pH the equilibrium is shifted to the reduced, -SH form, under more basic conditions the -SH group is more willing to be oxidized and replaced by -SR, where R is anything except for hydrogen).
CONTRIBUTION OF CYSTEINE IN PRIMARY STRUCTURE IN PROTEINS
According to the conventional Linderstrom-Lang criteria (Linderstrom-Lang, 1952; Linderstrom-Lang & Schellman, 1959) we can specify four general levels of protein structure:
- primary structure
- secondary structure
- tertiary structure
- quaternary structure
In this hierarchy primary structure is defined as the number and sequence of amino acid residues linked together by peptide bonds, from N-terminus to C-terminus.
At present there are listed six general levels of protein structure (Cantor & Schimmel, 1980; Creighton, 1993) :
- primary structure
- secondary structure
- supersecondary structure
- domains
- tertiary structure
- quaternary structure
where primary structure concerns amino acid sequence including post-translational modifications and disulfide bridges. The meaning of primary structure was then extended to topology of disulfide bridges in proteins.
The topology pattern of disulfide bridges is a very useful tool for identification protein families and superfamilies with respect to their common ancestry as well as their biological function.
A example of this is presented on Fig. 3 where proteinase inhibitors from different biological sources were divided into groups of identical disulfide topology. This is especially useful in searching the related proteins of low level of overall homology in primary structure.
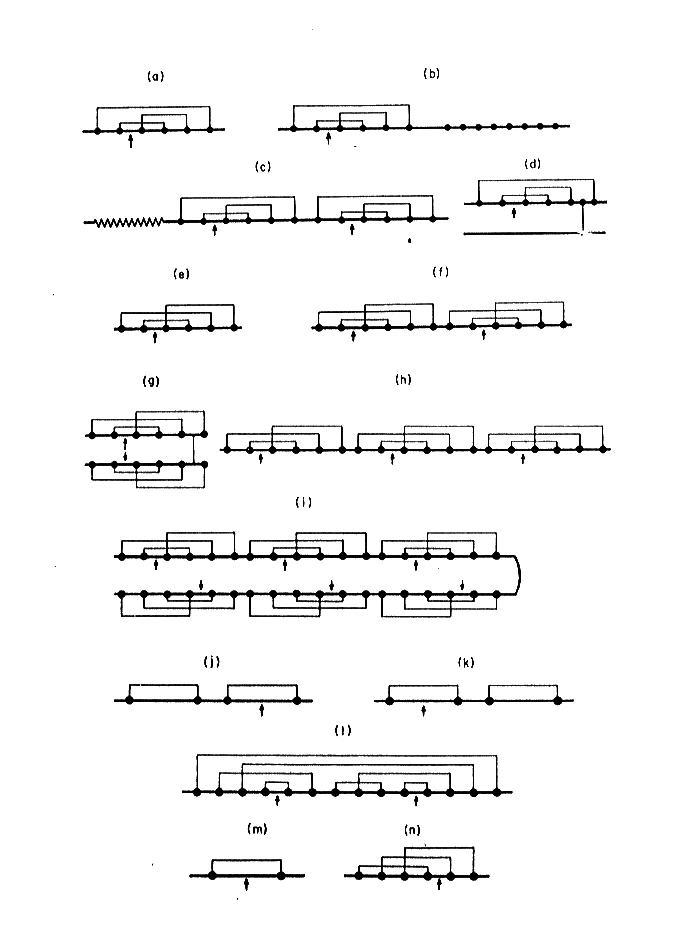
Fig. 3. Topology of disulfide bridges within selected proteinase inhibitory families. Solid circles indicate cysteine residues, and arrows the reactive sites. The schematic representations refer to the inhibitors that belong to: a-d) Kunitz family (BPTI-type), e-i) Kazal family (PSTI-type), j) inhibitor from Streptomyces, k) Kunitz-type family from soybean, l) Bowman-Birk family, m) potato type I family, n) potato type II family
Cysteine residues in homologous proteins (those of common ancestry) are very conservative according to PAM 250 scoring matrix (Fig 4). Only Trp is statistically more conservative than Cys.
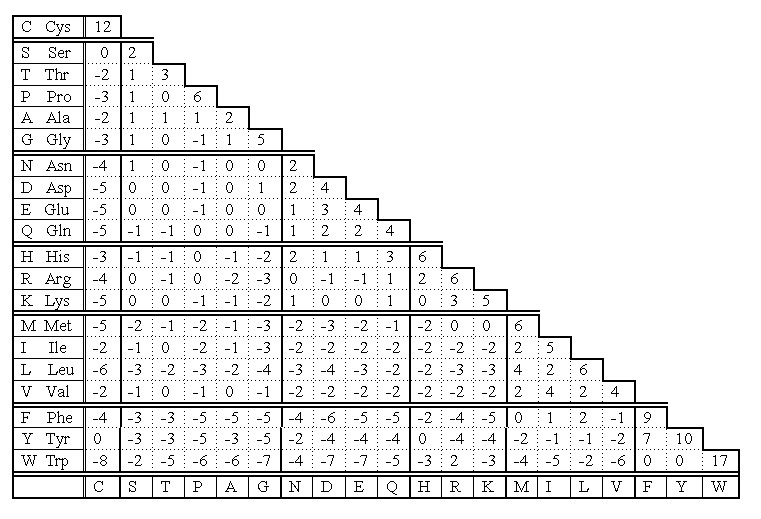
Fig 4. PAM 250 scoring matrix of Dayhoff mutation data matrix. Note that the highest values indicating replacement of a residue into itself is for Trp and Cys. That means the two residues are the most conservative in homologous sequences.
The reason is that cysteine - because of disulfide bridges - plays very important role in stabilization of protein structure at higher level. Its replacement would be dramatic for the overall structure of entire protein. If PAM 250 scoring matrix was set on different basis, the cysteine would be the most conservative residue in primary structure of homologous proteins. We could obtain such result if we count separately cysteines and cystines in this scoring. Thus, the cysteine takes the second place in its conservativity, because its score is the mean score for all cysteine residues regardless of their involvement to the formation of disulfide bridges.
Taking under consideration the conservative "behavior" of cysteine, the analysis of homology and alignment of related sequences usually starts with searching cysteines and locating them at appropriate positions - opposite to each other. This helps to locate properly the gaps resulting from insertion/deletion of the other amino acid residues (Fig 5).

Fig. 5 Multiple alignment of cysteine-rich squash proteinase inhibitors. The arrow marks the reactive site peptide bond of the inhibitors. The positions which are 100% conservative are framed. Note that all cysteines are 100% conservative.
CONTRIBUTION OF CYSTEINE IN SECONDARY STRUCTURE IN PROTEINS
Secondary structure of proteins is dependent on amino acid sequence. In that effect there are included side chains of amino acids and periodicity of their occurrence along the polypeptide chain. For that reason we can admit some residues as helix stabilizers or helix "breakers", etc.
For helix formation it is important that every 4th residue is of the same character according to it's hydrophobicity, and sterically acceptable (no steric hindrance). Strongly hydrophobic amino acids, such as Leu, Ile, Val are considered to be helix stabilizers in many prediction algorithms. Cysteine which is not involved in disulfide bridge formation can also stabilize alpha helix, although high reactivity of -SH group can possibly distort the regular structure by interaction with the other reactive groups. The situation can dramatically change when cysteines in helical fragment form disulfide bridges. This may imply conformational changes which don't allow the fragment to accept the helical regularity since disulfide bridges are stronger than the forces stabilizing alpha helix.
As an example let's consider two small proteins representing opposite situations.
One of them is transcription associated Rop protein (represor of primer fro E.coli) (Fig. 6) which has two cysteine residues not involved in S-S formation. The other one is CMTI-I (Fig. 7), the trypsin inhibitor from squash, containing six cysteine residues which form three disulfide bridges.
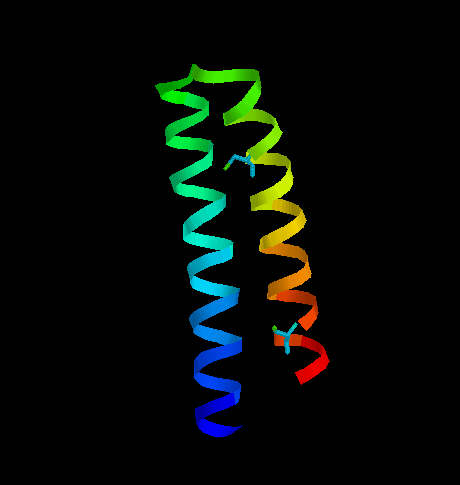
Fig. 6. Transcription associated Rop protein from Escherichia coli. The two cysteine residues (not involved in S-S formation) are shown.
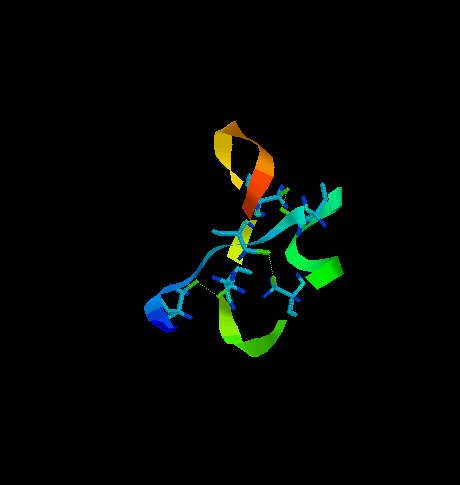
Fig. 7. The squash trypsin inhibitor from Cucurbita maxima (CMTI-I). Six cysteines forming three disulfide bridges are shown. Note the irregularity in secondary structure.
Rop protein has a very regular structure, it consists of two alpha helices linked by turn. the only two cysteine residues are located far from each other and do not interact with each other.
CMTI-I does not show any long helical fragment. The entire secondary structure is rather irregular and very rigid because of disulfide clips. It is important to note that the statistical algorithm of Garnier et al. (1978) predicts for CMTI-I significant contribution of helical fragments (Fig. 8 and 9) which is absolutely not true. The reason of such inconsistency is that the algorithm does not recognize the difference between cysteine and cystine and does not take the effect of S-S bridges on secondary structure under consideration. Probably, if all cysteine residues were in the reduced form (with free sulfhydryl groups) high percentage of helical fragments would be possible.
Helix Decision Constant = -100
Extended Decision Constant = -75
Turn Decision Constant = 0
coil Decision Constant = 0
Amino Acids in Helix Conformation H = 20 69.0%
Amino Acids in Extended Conformation X = 3 10.3%
Amino Acids in Turn Conformation T = 6 20.7%
Amino Acids in coil Conformation C = 0 0.0%
RVCPRILMECKKDSDCLAECVCLEHGYCG
XXTHXHHHHHHHHHHHHHHHHHHHTTTTT
Fig. 8. Garnier Secondary Structure Prediction for CMTI-I. The results obtained from program PROTSE.
predict7.htm - click here to see the results
Fig. 9. Garnier Secondary Structure Prediction for CMTI-I. The results obtained from program PREDICT7 (Carmenes et al., 1989).
Of course similar effects of disulfide "clipping" on secondary structure can be expected in case of every type of secondary structure. Therefore, algorithms which do not recognize cysteine and cystine as separate units, cannot have high predictive accuracy, and may give results far from the actual situation.
CONTRIBUTION OF CYSTEINE IN TERTIARY AND QUATERNARY STRUCTURE IN PROTEINS
The role of cysteine in tertiary structure of proteins is obvious. The disulfide bridges formed by these residues link the fragments within a polypeptide chain, sometimes located very far from each other with respect to their primary structure. Thus, the cysteine residues play the crucial role in the final three dimensional conformation of the protein molecule, and make it stable (see Fig. 7). Disulfide bridges are also involved in the formation of the loops of the backbone. These loops can be exposed on the surface of the molecule (far from the core) and available for the other factors to interact with them. This is important for the fragments containing residues involved in recognition mechanism. Disulphide bonds improve rigidity of the protein. It has very significant meaning in cysteine-rich proteins such as proteinase inhibitors, where even cleavage of the reactive site peptide bond doesn't change it's overall conformation and such 'modified' inhibitor still possesses antiproteinase activity (Fig. 7).
The loop formation stabilized by disulfide bridges can be observed in many different proteins. An example of that are immunoglobulins, in which light and heavy chains have two and four such loops respectively. They are recognized as repetitive domains in this class of proteins, and therefore they are classified as mosaic proteins.
The quaternary structure practically exists thanks to presence of cysteine related disulfide bridges linking separate polypeptide chains. In most proteins which consist of more than one polypeptide chain, the chain are kept together by interchain disulfide bridges. The simple example is insulin, where A and B chain are linked by two bridges between CysA7 - CysB7 and between CysA20 - CysB19. Another example is chymotrypsin. This enzyme is converted from its zymogen by limited proteolysis in a few places. Chymotrypsynogen consists of a single long polypeptide chain, the active enzyme contains two or three chains, according to the step of activation. These chains of active enzyme could not exist together without cysteine disulfide bridges linking them. Generally we could say then, that without cysteine residues quaternary structure wouldn't exist.
CYSTEINE IN BIOLOGICALLY ACTIVE OLIGOPEPTIDES AND PROTEINS
Except for the structural properties of proteins, cysteine residues may also play important role in catalytic processes of enzymes, in storage and adjusting the reduction potentials, they can be a source of sulfur assimilable in easy way, etc.
A good example of the essential role of cysteine in enzyme catalytic activity are thiol proteinases. This is one of four major classes of proteinases. To this class belong such proteinases as bromelain, papain, ficin and some cathepsins. The common feature of these enzymes is that they all have cysteine residue as the central amino acid of catalytic site (Fig. 10). Cysteine is directly involved in the formation of acyl intermediate with the substrate.
The overall mechanism of catalysis reaction by thiol proteinases is the same as in serine proteinases. Cysteine at position 25 in papain has the same function as serine at position 195 in trypsin, chymotrypsin or other serine proteinases. The transition state in catalysis by papain is shown on Fig. 10.
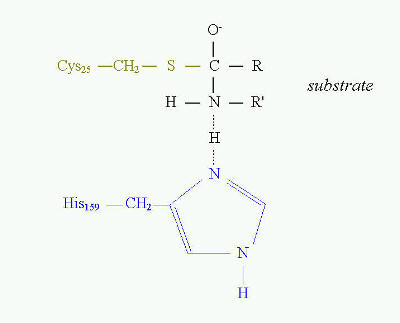
Fig. 10. Transition state in proteolytic catalysis by papain. The -SH group of Cys25 participates in the formation of acylenzyme intermediate with the substrate (shown in black). His159 serves as proton acceptor, which interacts directly with nitrogen of the peptide bond to be cleaved. Sulfhydryl group of catalytic Cys25 of papain acts similarly to Ser 195 in serine proteinases.
The different role of cysteine is in tripeptide - glutathione (Fig. 11). It is present in the equilibrium of two forms - reduced and oxidized. The reduced form serves as "sulfhydryl buffer" that maintains the cysteine residues of hemoglobin and other erythrocyte proteins in the reduced state. It also works as detoxitant by reacting with hydrogen peroxide and organic peroxides. It protects red cells from oxidative damage. Glutathione is involved in maintaining the proper equilibrium of the reduced form of the other reduction potential carrier - NADPH. Thus, the presence and state of glutathione is essential for the activity of many dehydrogenases (for example glutathione reductase).
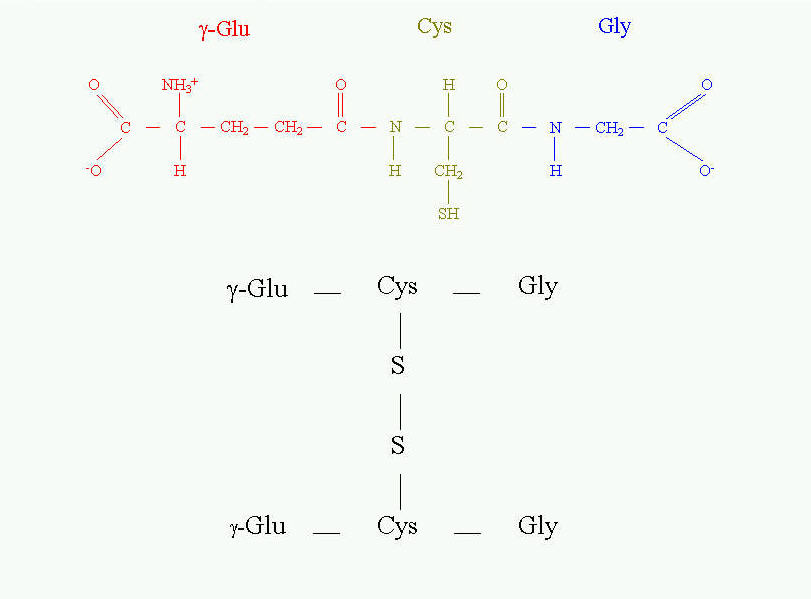
Fig. 11. Glutathione and its oxidized form.
SO WHY CYSTEINE IS SPECIAL?
REFERENCES:
Written by:
Institute of Biochemistry and Molecular
Biology
University of Wroclaw
50-137 Wroclaw
Tamka 2
Poland
e-mail: lulu@bf.uni.wroc.pl