
|
Site Directed Mutagenesis of Tyrosyl tRNA Synthetase |
Protein synthesis occurs at the ribosome via the stepwise condensation of amino acids. Correct incorporation of amino acids is brought about by transfer RNA molecules (tRNA) charged with amino acids. Individual tRNA's contain a recognition element (the anticodon loop) which recognises the complementary triplet of bases on the m-RNA encoding for the amino acid in the protein, so ensuring incorporation of the correct amino acid. A precursor step to this process is the formation of the amino acid-tRNA molecule, which is catalysed by the appropriate amino acid tRNA synthetase. In the case of tyrosyl-tRNA synthetase from Bacillus stearothermophilus the enzymatic function has been studied extensively using a combination of X-ray crystallography, enzyme kinetics, site directed mutagenesis and molecular modelling to elucidate the roles played by key amino acid residues in the catalytic function of the enzyme.
Tyrosyl tRNA synthetase from Bacillus stearothermophilus is a dimeric enzyme, consisting of two identical sub-units (MW 2 x 47.5 kDa). It catalyses the formation of tyrosyl-tRNA in a two step reaction [1] (Figure 1). In this reaction, tyrosine is first activated by reaction with ATP to form the enzyme bound intermediate, tyrosine adenylate, with the liberation of pyrophosphate. The amino acid is then transferred to the 3' adenosine of tRNATyr with the release of AMP. Although the enzyme is dimeric, only 1 molecule of tyrosine is bound per dimer, resulting in only 1 molecule of product per dimer.

Figure 1: Catalysis of Tyrosyl tRNA formation
The progress of this reaction can be readily studied using filter binding assays with radiolabelled substrates.
A key feature of this system that has made it particularly amenable to study is the stability of the tyrosyl adenylate intermediate formed in step 1. If the enzyme is incubated with tyrosine and ATP in the presence of inorganic pyrophosphatase, which hydrolyses the pyrophosphate formed in step 1, then the reverse reaction is blocked and the enzyme-tyrosyl adenylate complex is formed stoichiometrically. This has two important consequences:
1. It allows the concentration of active sites to be measured precisely using the radioactive assay. This is particularly important when comparing data from mutant enzymes since changes in Vmax must be caused by changes in Km and kcat and not for the trivial reason that there is a change in the proportion of enzyme which remains functional.
2. It is possible to crystallise the intermediate complex and determine its structure by X-ray crystallography.
A third factor that has enabled detailed study of tyrosyl tRNA synthetase from Bacillus stearothermophilus is that the enzyme and mutants can be readily expressed to high level in E. coli and subsequently purified to homogeneity.
Crystal structures of tyrosyl tRNA synthetase from Bacillus stearothermophilus have been solved for the enzyme itself, the enzyme complexed to tyrosine and to tyrosyl-adenylate intermediate and for several mutants [2-6]. Each monomer of the enzyme has three domains: an alpha/beta domain containing a six stranded beta sheet (residues 1-220) , an alpha-helical domain (residues 248-318) and a disordered C-terminal domain (residues 319-418), Deletion mutagenesis has shown that the N-terminal 319 residues is primarily responsible for the activation reaction [7] (step1, Figure 1), while the disordered C-terminal domain is largely responsible for tRNA binding. The crystal structure of the truncated N-terminal domain is very similar to that of the wild type enzyme [4]
Click here to view the structure of the enzyme complex with tyrosyl-adenylate intermediate with rasmol. The structure reveals eleven key hydrogen bond interactions between enzyme and the tyrosyl-adenylate intermediate. The residues involved can be viewed by applying the following rasmol script.
restrict ligand, 34, 35, 36, 38, 48, 51, 169, 192, 195
zoom 300
These interactions are shown diagramatically below (Figure 2).

Figure 2: Schematic Representation of Hydrogen Bonding of Tyrosyl Adenylate Intermediate [8]
The structure of the enzyme complexed to the reaction intermediate [2,8] allowed the the relative importance of each of these hydrogen bonding interactions to be assessed using site directed mutagenesis.
The first residue to be mutated was Cys-35 [9, 10] which contacts the sugar of the ATP and therefore plays a role in substrate binding, but is distant from the site where the chemical reaction occurs (i.e. at the phosphate group). Cys-35 was mutated to both Gly and Ser, to test the effect of deleting the side chain hydrogen bonding interaction in the case of Gly or replacing it by an alternative amino acid capable of offering an alternative hydrogen bond (-OH on Ser side chain). Both substitutions resulted in enzymes which operated with similar but lower efficiencies than the wild type enzyme. This reflected a decrease in the binding energy in the transition state, by ca 1.2 kcal /mol. In the case of the Gly mutation this can be easily explained by the loss of a hydrogen bond to the substrate caused by the deletion of the Cys. At first sight, this might lead to the expectation that the Ser mutation would lead to the opposite phenomenon than that observed (i.e. increased substrate binding) since the more polar hydroxyl group in Ser can form stronger hydrogen bonds than those formed by the -SH group in the native Cys. The observed loss of binding energy in the Ser mutant has been explained by considering the interactions of enzyme, substrate and solvent. In the absence of substrate the active site of the enzyme is solvated by water and the hydrogen bonds formed must be sacrificed as the substrate binds. Also, due to the smaller size of an oxygen atom compared with sulphur, the Ser OH falls short of the position of the Cys SH by about 0.5 angstroms, requiring structural rearrangement to form a good hydrogen bond. These factors combine to make the formation of a hydrogen bond from Ser to the sugar hydroxyl group energetically unfavourable, since Ser must sacrifice a strong hydrogen bond to solvent for a relatively weak bond to the substrate. In fact, since the Km and kcat of the Gly mutant enzyme are essentially the same as that for the Ser mutation, it seems unlikely that a hydrogen bond from Ser-35 play any part in hydrogen bonding to the ribose group on the substrate. Initially, this seemed to account well for the facts, however a subsequent higher resolution crystal structure has shown this explanation to be only partially correct. In the higher resolution structure, the sulphur-ribose oxygen distance has been found to be too long for an effective hydrogen bond to be formed to the ribose 3'-OH. In fact the contact is bridged by a water molecule, so that the cysteine side chain hydrogen bonds to water which in turn hydrogen bonds to the ribose hydroxyl. Presumably, in the Ser mutant, the distance between the Ser hydroxyl and ribose hydroxyl is too great to be bridged by a water molecule. Other residues making hydrogen bond contacts with the tyrosyl adenylate intermediate have been systematically mutated and analysed kinetically. The results are summarized in table 1 [8].
| Comparison | |||
| Enzyme 1 | Enzyme 2 | Substrate |  kcal/mol kcal/mol |
| Phe-34 | Tyr-34 | Tyr | 0.52 |
| Gly-35 | Cys-35 | ATP | 1.14 |
| Ala-51 | Cys-51 | ATP | 0.47 |
| Gly-48 | Asn-48 | ATP | 0.77 |
| Gly-48 | His-48 | ATP | 0.96 |
| Ser-35 | Cys-35 | ATP | 1.18 |
| Phe-169 | Tyr-169 | Tyr | 3.72 |
| Gy-195 | Gln-195 | Tyr | 4.49 |
| Gly-35 | Ser-35 | ATP | -0.04 |
| Ala-51 | Thr-51 | ATP | -0.44 |
Table 1: Relative binding energies of groups in tyrosyl tRNA synthetase inferred from comparisons between mutant and wild-type enzyms at 298K [8]
The final column in the above table reflects the change in free energy of the transition state between the wild type and mutant enzymes. This can be calculated from the equation shown below in Figure 4 which shows the energy changes that occur as the substrate binds to the enzyme and passes to the transition state (highest energy point) in the reaction [10] .
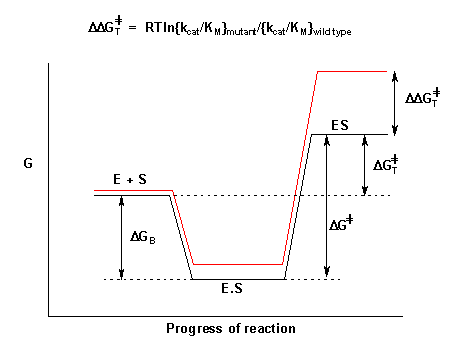
Figure 3: Comparison of reaction profile of wild type and mutant enzyme
The effects of the mutations described in table 1 fall into two categories:
(i) deletion of a hydrogen bonding interaction between uncharged groups on substrate and enzyme, resulting in a change in free energy of the transition state of 0.5-1.5 kcal/mol;
(ii) deletion of a hydrogen bond interaction between the enzyme and a charged group on the substrate, resulting in a loss of binding energy of 4 kcal/mol.
These changes translate into rate differences (kcat/Km) of 2-12 fold in the former case while the latter can give rise to a 1000-fold difference in rate. An explanation for the differences in the magnitude of the two effects can again be offered by a consideration of the role of the solvent in the binding process.
Case 1: Deletion of of H-bond between uncharged groups
The change in hydrogen bonding patterns in moving from the wild type and mutant enzyme can be summarized by the following two equations:

Although a change in the number of hydrogen bonds changes in moving from the wild type to the mutant enzyme, the same number and type of hydrogen bonds are broken as those formed. Very little change in free energy of binding would therefore be expected from this type of mutation. What small changes are observed can only be accounted for by a more detailed consideration of the exact nature of the hydrogen bonds involved (as in the case of the Cys-35 mutations described above). A similar situation occurs when a charged group on the enzyme which hydrogen bonds to the substrate is deleted:

In the mutant the charge-dipole interaction is lost from both sides of the equation, so the change would be expected to confer little change on binding energy. This is observed in the loss of the interaction of His-48 with the ribose ring oxygen, which shows only a 0.5 kcal/mol loss in binding energy.
Case 2: Deletion of H-bond with charged group on the substrate
This can be summarized by the following equations:

In this case, for the mutant, the charge-dipole interaction on the left hand side is counterbalanced only by a dipole-dipole interaction on the right hand side. In binding substrate and the transition state, the mutant enzyme creates unsolvated charge, which has a high energy penalty.
In summary, these studies have shown that hydrogen bonds generally confer small binding energies, except where they solvate charged groups. Examples are Tyr-169, which interacts with the tyrosine ammonium group of the substrate and Gln-195 which probably interacts with the tyrosine carboxylate group in the transition state. This effect can also be invoked to account for the high selectivity of tyrosyl tRNA synthetase for tyrosine over phenylalanine. The two amino acids differ only by the presence of a phenolic hyroxyl on the tyrosine ring:

In Figure 2, Asp-176 carboxylate side chain hydrogen bonds to the tyrosine -OH. Phenylalanine is not capable of providing a similar interaction, so in binding to the active site would leave the Asp-176 side chain without a hydrogen bonding partner. From above discussion, this results in a large energy penalty and is the basis for effective discrimination between the two amino acids
From the crystal structure of tyrosyl-tRNA synthetase with the tyrosyl adenylate intermediate it has been possible to identify key residues involved in substrate binding and assess the nature of their contributions using site directed mutagenesis. Unfortunately the beta and gamma phosphate groups of ATP in step 1 of the reaction are not present in the crystal structure, and no structural information is therefore available on how exactly the enzyme brings about the large rate enhancement in tyrosyl adenylate formation that is observed. An alternative approach based on molecular modelling has been used to help determine which other residues are involved in the catalytic action of the enzyme.
If we consider the reaction mechanism for formation of the tyrosyl adenylate intermediate, the reaction proceeds through a penta-coordinate transition state arising from nucleophilic attack of the tyrosine carboxylate at the alpha-phosphate group [11]. The expected transition state has been modeled into the crystal structure (Figure ) and shows that the beta and gamma phosphate groups of ATP are able to make stabilising charge-dipole contacts with the side chains of Thr-40 and His-45 [12-14].

Figure 4: Model of transition state for penta-coordinate transition state in tyrosyl adenylate formation
The importance of these contacts has been confirmed by site directed mutagenesis, where mutations to Ala-40 and Gly-45, resulted in 7000- and 200-fold reductions in rate. The double mutant (Ala-40 and Gly-45) showed a dramatic 300,000 fold reduction in rate.
Other residues have also been found to play key roles in catalytic activity, but their role is not immediately obvious from the model since they are either remote from the bound reagents or so disordered that they are difficult to localise in the crystal structure. The identification of these residues relied upon a more empirical approach where residues with charged side chains were randomly mutated [15]. Arg-86 and Lys-230 have been implicated in binding ATP in the transition state and pyrophosphate in the [Enzyme-Tyr-AMP.PPi] complex, while Lys-82 and Lys-233 have similar interactions but also bind ATP in the [E.Tyr.ATP] complex. The side chains of Lys-82 and Arg-86 have high B-factors (i.e are mobile) but are within hydrogen bonding distance of the beta and gamma-phosphate oxygens in the transition state model. Lys-230 and Lys-233 are so disordered that they cannot be located in the X-ray map. However, even when modelled in their extended conformations the side chain amino functions cannot be located closer than 8 angstroms from the phosphates of ATP in the transition state, suggesting that the loop containing these residues must move to wrap around the transition state in an induced fit mechanism. Table 2 summarises the interaction energies of the side chains of tyrosyl tRNA synthetase with the various reaction components [16].
| Tyr | ATP | [Tyr-ATP] | PPi | Tyr-AMP | |
Tyrosine Binding Site | |||||
Tyr-34 Asp-78 Tyr-169 Gln-173 | + + + + + + + + + + + + + | 0 + +* 0 + +* | + + + + + + + + + + + + + |
0 + +* 0 +* | + + + + + + + + + + + + + |
Nucleotide and Pyrophosphate Site | |||||
Cys-35 Thr-40 His-45 His-48 Thr-51 Lys-82 Arg-86 Asp-194 Lys-230 Lys-233 | 0 0 0 0 0 0 0 0 0 0 | 0 0 0 0 0 + + 0 0 0 + + + + | + + + + + + + + + + + + + 0 + + + + + + + + + + + + + + + + + + + + |
0 + + + + + + + + 0 0 + + + + + + + + + + + + + + + + + | + + + 0 0 + + + - 0 - + + + 0 0 |
| Apparent stabilisation energy from side chain in kcal/mol: 0=-0.5 to +0.5; + = 0.5-1.0; + + = 1.0-1.5; + + + = 1.5-2.0; + + + + = >2.0; - = -0.5--1.0; * = evidence for structure disruption on mutation | |||||
Table 2: Interaction Energies of Side Chains of Tyrosyl tRNA Synthetase with Reagents [16]
Two of the residues in table 2, Lys-230 and Lys-233, form part of the signature sequence K-M-S(T)-K-S(T) present in class I aminoacyl-tRNA synthetases [17]. Thr-234 from this sequence has also been shown by site-directed mutagenesis [18-21]to be an important residue in the catalytic activity of tyrosyl tRNA synthetase. Mutation to alanine reduces the forward rate constant for formation of tyrosyl adenylate 500-fold, while mutation to serine results in only a 4-fold decrease, suggesting that the loss of the hydroxyl group in the T234A mutant is responsible for the decreased reaction rate. The stabilising effect of the hydroxyl group on the tyrosyl adenylate transition state is 2.7 kcal/mol, equivalent to the energy of disrupting a hydrogen bond involving a charged species (see above). This suggests that Thr-234 may form a hydrogen bond to one of the phosphate groups on ATP, as has been proposed for Lys-230 and Lys-233. Alternatively, it may reflect that Thr-234 is coordinated to the Mg2+ ion bound to ATP. This has been investigated by replacing the magnesium ion with cadmium. In the wild type enzyme, this had no effect on the stability of the transition state complex E.[Tyr-ATP], while in the T234A, the complex is less stable when cadmium is used in place of magnesium. From this analysis it is clear there is some coupling between Thr-234 and metal ion but it has not been determined whether this is a direct or indirect effect. Double mutant experiments, mutating Lys-230 to alanine, together with Thr-234 mutations have shown that the interactions of Thr-234 and Lys-230 are in some way coupled, since the effects of the double mutation are not additive, although again the nature of the interaction has not been determined.
From the above discussion it is clear that the first step in the catalytic action of tyrosyl tRNA synthetase, formation of tyrosyl adenylate, has been studied in great detail and is now perhaps one of the best characterised enzymatic reactions. By comparison, the second step in the process, the formation of tyrosyl tRNA is poorly understood. Deletion mutagenesis has strongly implicated the disordered C-terminal domain in tRNA binding [7]. When this domain is truncated (leaving residues 1-319), the fragment is nearly kinetically identical with the wild type enzyme in the activation step but is unable to bind or aminoacylate tRNA.
Further studies using the truncated domain in heterodimers with full length enzyme have proved invaluable for elucidating the mode of binding of tRNA. For example, the heterodimer between wild type and truncated subunit is only slightly disabled (Vmax reduced by a factor of 2 [22]. However, if His-45 is mutated to Asn in the truncated subunit the rate of formation of tyrosyl adenylate is reduced by 104. Putting this mutation into only the full length subunit has no effect on the rate of aminoacylation. This is illustrated below, and shows that although tRNA binds predominantly to the full length subunit, amino acid acceptance occurs from the small subunit.

Similar experiments have been carried out with heterodimers, where basic residues have been systematically mutated [15]. Basic residues carry a positive charge and they may be expected to interact with the many negatively charged phophate groups present in tRNA. The results of these experiments show that tRNATyr interacts with Lys-151, Arg-207 and Lys-208 in the small subunit, and six basic residues in the disordered C-terminal domain of the full length subunit.
A further aspect to the interaction with tRNA is that not only must the enzyme bind tRNA and carry out the chemical transformation, but it must also discriminate tRNATyr from the 19 other species of tRNA. High specificity in this reaction is crucial to allow faithful translation of the genetic code. It has been noted that when Glu-152 is mutated to Ala the resulting protein is toxic in the cells producing it, where no toxic effects are noted when the wild type protein is produced. This is due to erroneous interactions between tyrosyl t-RNA synthetase and non-cognate tRNAs [23]. This has been extended to the study of a range of mutations for Glu-152 [24]. The effect of these mutations was studied in vitro by the ability of the mutated protein to acylate tRNAPhe and tRNAVal and in vivo where production of the mutant protein was under the control of an inducible promotor (Ptac), so that the toxic effects of the mutants were not apparent until the promotor was induced. The misaminoacylations of tRNAPhe and tRNAVal with tyrosine in vitro and toxicity of the induced protein in vivo correlated with the change in nature of the side chain at position 152 from negatively charged to positively charged. Arg-152 resulted in the both the highest levels of misincorporation of tyrosine and highest levels of toxicity. This seems to indicate that Glu-152 exhibits a negative role in discrimination between tRNAs, mediating electrostatic repulsion of non-cognate tRNAs. The same mechanism appears to operate in other prokaryotic tyrosyl t-RNA's, where Glu-152 is either conserved or replaced by Asp.
1.R Jakes & AR Fersht (1975), Biochemistry, 14, 3344.
2. TN Bhat, DM Blow, P Brick & J Nyborg (1982), J. Mol. Biol, 158, 699.
3. P Brick, TN Bhat & DM Blow (1988), J. Mol. Biol., 208, 83.
4. P Brick & DM Blow (1987), 194, 287.
5. KA Brown, P Brick & DM Blow (1987), Nature, 326, 416-418.
6. MD Fothergill & AR Fersht (1991), 30, 5157.
7. MMY Waye, G Winter, AJ Wilkinson & AR Fersht (1983) EMBO J., 2, 1827
8. AR Fersht, J-P Shi, J Knill-Jones, DM Lowe, AJ Wilkinson, DM Blow, P Brick, P Carter, MMY Waye & G Winter (1985) Nature, 314, 235.
9. G Winter, AR Fersht, AJ Wilkinson, M Zoller & M Smith (1982), Nature, 299, 756.
10. P. Moody & AJ Wilkinson (1990), in Protein Engineering (Ed. D Rickwood), pp32-33, IRL Press.
11. G Lowe & G Tansley (1984), Tetrahedron lett., 40, 113.
12. RJ Leatherbarrow, AR Fersht, & G Winter (1985), Proc. Natl. Acad, Sci. USA, 82, 7840.
13. AR Fersht, TNC Wells & RJ Leatherbarrow (1986), Trends Biochem Sci, 11, 321.
14. RJ Leatherbarrow & AR Fersht (1987) Biochemistry, 26, 8524.
15. H Bedouelle & G Winter (1986), Nature, 320, 371.
16. AR Fersht (1987), Biochemistry 26, 8031.
17. C Hounrondji, P Dessen & S Blanquet (1986), Biochimie 68, 1071.
18. EA First & AR Fersht (1993), Biochemistry, 32, 13644.
19. EA First & AR Fersht (1993), Biochemistry, 32, 13651.
20. EA First & AR Fersht (1993), Biochemistry, 32, 13658.
21. EA First & AR Fersht (1995), Biochemistry, 34, 5030
22. P. Carter, H. Bedouelle & G. Winter (1986), Proc. Natl. Acad. Sci. USA, 83, 1189
23. A Vidal-Cross & H Bedouelle (1992), J. Mol. Biol., 223, 801
24. H Bedouelle & R Nageotte (1995), EMBO J., 14, 2945