THE FACTORS INVOLVED IN PROTEIN THERMOSTABILITY
Marialuisa Gazerro, PPS course participant, 1997
The question of why proteins are stable is of fundamental importance in biochemistry. It is central to understanding the relationship between amino acid sequence and three dimensional structure, and ultimately in the prediction of a protein's structure from its sequence.
Our knowledge of factors involved in protein stabilization (see Day's PPS Project) is usually based on results of research on organisms living in environments characterized by moderate conditions regarding temperature, pH, salinity, oxygen pressure, etc. These conditions are routinely considered as normal, neglecting the fact that presumably life originated under environmental circumstances far apart from those. Actually, a large number of microorganisms have been discovered within the past decade which are thriving in more or less bizarre habitats as deep seas black smokers, alkaline lakes, volcanic systems, and extremely acidic environments like puddles of solutions seeping from ores or mine-refuse piles. They are called extremophiles, which include extreme thermophiles, halophiles, acidophiles, alkalophiles, psychrophiles, osmophiles, barophiles, radiation resistant and heavy metal tolerant organisms. The majority of them belongs to a distinct branch of the universal tree of life which was added to the traditional division of the biological world into Prokaryotes and Eukaryiotes, and is called the third kingdom of Archaebacteria (Archaea). A diverse range of bacteria, cyanobacteria, algae and yeast have been isolated from the above-mentioned habitats and their proteins have been studied.
The comparisons of the sequences and crystal structures of proteins from extremophilic microorganisms with their corresponding counterparts from mesophilic organisms have given insight in the mechanisms that nature has employed to increase the stability of proteins. The extraordinary stability of extremozymes does not only provide suitable model systems to understand the factors involved in protein stability, but also open a wide field of microbiological and technical applications. Elucidating the mechanisms by which life persists under extreme conditions might suggest new approach to tackling problems in, for example, medicine, bioconversions or bioremediation of waste (Herbert, 1992; Schäfer, 1992).
This project focuses on protein thermostability and is intended to discuss the factors involved in protein thermostability recognized in hyperthermophiles, so far. Given the breadth of the subject area, this must of necessity be selective and can provide only a broad overview. For more detailed information, the reader should consult the references.
Contents Hyperthermophiles and their proteins
HYPERTHERMOPHILES AND THEIR PROTEINS
The temperature range of growth has been used to classify groups of organisms. The commonly used divisions are psychrophiles (-5 to +20°C), mesophiles (15-40°C) and thermophiles (45-100°C or more). Moderate thermophiles have maximal growth temperatures between 55 and 80°C. Hyperthermophiles (or extreme thermophile) are defined as organisms that grow optimally at 80°C or higher with a maximal growth temperature of 90°C or higher. Many believe that the maximum growth temperature for organisms as we know them will be found to be between 120° and 150°C. The ultimate limit of growth at temperatures beyond 120°C must be ascribed first to the susceptibility of the covalent structure of biopolymers towards hydrolysis and hydrothermal degradation and second to the fact that hydrophobic interactions seem to vanish close to this temperature limit.
Hyperthermophiles (Baross et al., 1996) were first isolated more than 20 years ago. Most hyperthermophiles that have been isolated so far belong to the phylogenetical domain of Archaea, whereas only two families, the isolates of Thermotogales and Aquifex, belong to the kingdom of Bacteria.
At present, approximately 20 genera and more than 40 species have been described. Phylogenetic schemes based on ribosomal RNA and selected protein sequences point to hyperthermophiles as the most ancient of extant organisms and perhaps the most slowly evolving organisms, further implying a hyperthermophilic ancestor of all life. Their source environment include terrestrial and shallow- and deep-marine volcanic and geothermal systems. Several genera of hyperthermophiles are capable of growth at a pH as low as pH 0.5. Marine isolates require NaCl for growth, and deep-sea species grow and survive best at their higher growth temperatures under hydrostatic pressure equivalent to or higher than the in situ pressures of their environments (Michels et al., 1996). The vast expanses of deep-sea geothermal environments have only begun to be explored, and the future promises the discovery of new organisms capable of growing at even higher temperatures and pressures. Most of these organisms are anaerobes and include chemoorganotrophs, chemolithotrophs, and methanogens. Many employ novel metabolic pathways, have extraordinary heat-stable proteins and use ingenious strategies for stabilizing nucleic acids and other macromolecules in vivo. In most cases, adaptation to thermal stress in the course of evolution resulted in thermophily rather than thermotolerance. This means such organisms require high temperature for their life cycle: they are unable to grow or multiply under mesophilic conditions.
Coping with extreme condition the cellular components may exhibit high intrinsic stability or the cells may provide extrinsic factors (such as ligands, chaperones, etc.) or increased synthesis in order to compensate for destruction. This project deals with factors involved in intrinsic thermostability of proteins.
Proteins from hyperthermophiles commonly exhibit high intrinsic thermostability. They are stable over an anomalously wide range of temperatures without undergoing heat or cold denaturation. This holds for the native protein in situ, as well as for the recombinant superstable proteins in a mesophilic host and in vitro. The increased rigidity of thermophilic proteins compared to their mesophilic counterparts becomes evident from a wide variety of observations, such as decreased H-D exchange rates of amide protons, enhanced activity at low denaturant concentrations, and increased resistance against proteolysis and denaturants.
The thermophilic proteins are closely similar to their mesophilic counterparts regarding basic topologies and enzyme mechanisms. They have maximum enzymatic activities near the optimal growth temperatures of the organisms. Their high intrinsic stabilities become marginal at their respective maximal growth temperatures, leading to similar conformational flexibility for homologous enzymes under their respective physiological conditions. Adaptation of biomolecules to extremes of physical conditions tends to maintain "corresponding states" regarding overall topology, flexibility and solvation, shifting the normal characteristics to the respective physiological conditions.
The building blocks of proteins from hyperthermophiles, as well all other extremophiles, are exclusively the canonical natural amino acids (information on these from the PPS course). Thus, molecular adaptation depends exclusively on mutational changes in their local and global distribution. The correlation of sequence alterations with changes in thermal stability is highly complex. The increments of stabilization observed for single, double, and multiple point mutations are approximately additive, corroborating the idea that thermal stability is the cumulative effect of small improvements at various locations within the core of the protein or between its subunits.
Contents Introduction Protein thermostability
The term protein thermostability refers to the preservation of the unique chemical and spatial structure of a polypeptide chain under extremes of temperature conditions. The molecular basis of thermal stability of globular proteins is a highly significant yet unsolved problem. In recent years a considerable effort has been made to understand the mechanisms that determine the thermal stability of proteins (Jaenicke et al., 1996).
In general, the conformational stability of a protein is defined as the free
energy change, ![]() G, for the reaction
folded
G, for the reaction
folded ![]() unfolded under physiological
conditions. Common cellular proteins exhibit only marginal stabilities, with
free energy of stabilization
unfolded under physiological
conditions. Common cellular proteins exhibit only marginal stabilities, with
free energy of stabilization
![]() GN
GN![]() U of the order of 50 kJ/mol, attributable
to the cumulative effect of attractive and repulsive interactions at many
locations within a given molecule. Two classes of noncovalent interactions,
electrostatic and hydrophobic, are involved in the formation of the native
secondary structure and the packing of the hydrophobic inner core
(Pace et al., 1996). Electrostatic interactions
(see
the PPS course) include ion pairs, hydrogen bonds, weakly polar interactions,
and van der Waals forces. Hydrophobic interactions
(information
at this link) imply van der Waals forces and hydration effects of nonpolar
groups. The contributions of these interactions compete with the decrease
in configurational entropy and repulsive forces resulting from space-filling
properties and charges, such that in total the free energy of stabilization
represents a small difference between large numbers. A few interactions added
to the large number of stabilizing and destabilizing interactions would suffice
to yield the small net energy difference required for thermostability. The
biological significance of the low
U of the order of 50 kJ/mol, attributable
to the cumulative effect of attractive and repulsive interactions at many
locations within a given molecule. Two classes of noncovalent interactions,
electrostatic and hydrophobic, are involved in the formation of the native
secondary structure and the packing of the hydrophobic inner core
(Pace et al., 1996). Electrostatic interactions
(see
the PPS course) include ion pairs, hydrogen bonds, weakly polar interactions,
and van der Waals forces. Hydrophobic interactions
(information
at this link) imply van der Waals forces and hydration effects of nonpolar
groups. The contributions of these interactions compete with the decrease
in configurational entropy and repulsive forces resulting from space-filling
properties and charges, such that in total the free energy of stabilization
represents a small difference between large numbers. A few interactions added
to the large number of stabilizing and destabilizing interactions would suffice
to yield the small net energy difference required for thermostability. The
biological significance of the low
![]() Gstab is the requirement
for balance between rigidity as a prerequisite of specificity, on one hand,
and flexibility in connection with binding, activation, and release of
substrates, etc., on the other. The functional state of a protein is
characterized by a well-balanced compromise of stability and flexibility.
Dramatic changes in stabilization energies do not occur even in the most
extreme environments. With a stability increment per residue one order of
magnitude below the thermal energy, the overall stability of a polypeptide
chain must involve cooperativity. Otherwise, the addition of amino acid
increments in the process of structure formation would not allow the thermal
energy to be overcome.
Gstab is the requirement
for balance between rigidity as a prerequisite of specificity, on one hand,
and flexibility in connection with binding, activation, and release of
substrates, etc., on the other. The functional state of a protein is
characterized by a well-balanced compromise of stability and flexibility.
Dramatic changes in stabilization energies do not occur even in the most
extreme environments. With a stability increment per residue one order of
magnitude below the thermal energy, the overall stability of a polypeptide
chain must involve cooperativity. Otherwise, the addition of amino acid
increments in the process of structure formation would not allow the thermal
energy to be overcome.
This is in keeping with the equilibria curves for the thermal denaturation of proteins. One characteristic of many such equilibria is their all or none character. In a very narrow temperature range, the native protein melts to a random coil. This is to be expected for a cooperative process, which sums a large number of small contributions. In terms of the thermodynamics, if we write an equilibrium constant for the thermal transition from the ordered to the random-coil form as:
k = fraction of residues in random coil regions/fraction of residues in ordered regions
we can say that
k = exp(-![]() G/RT) =
exp(-(
G/RT) =
exp(-(![]() H -
T
H -
T![]() S)/RT)
(1)
S)/RT)
(1)
where ![]() G is the free energy change
for the thermal transition of one residue from an ordered region to a random
coil one.
G is the free energy change
for the thermal transition of one residue from an ordered region to a random
coil one.
The transition can be abrupt only if
![]() H and
H and
![]() S are both large, so that the
exponent in equation (1) changes from a very large negative quantity to a
very large positive quantity for a small change in temperature. Now,
S are both large, so that the
exponent in equation (1) changes from a very large negative quantity to a
very large positive quantity for a small change in temperature. Now,
![]() H and
H and
![]() S for the loosening of a single
residue from an ordered region are modest quantities. Such values would yield
a gradual transition over the whole temperature range. But if the process
is cooperative, then the
S for the loosening of a single
residue from an ordered region are modest quantities. Such values would yield
a gradual transition over the whole temperature range. But if the process
is cooperative, then the ![]() H and
H and
![]() S would be the sum of all of
the contributions from individual residues and the change would be of the
type two-state transition. That means the process must be highly cooperative:
the entire chain (or large segments thereof) must change directly from the
ordered to the random-coil form.
S would be the sum of all of
the contributions from individual residues and the change would be of the
type two-state transition. That means the process must be highly cooperative:
the entire chain (or large segments thereof) must change directly from the
ordered to the random-coil form.
Various approaches are used to study the structural basis of thermostability. A very promising approach is the investigation of the structure-function relationship of homologous enzymes (see the PPS course) from mesophilic and thermophilic sources (Kelly et al., 1993; Tanner et al., 1996). The notion that increased thermostability results from an amalgam of many small changes throughout the enzyme is the basis for analyses such as calculating the total number of salt links, H-bonds, buried surface area, and surface to volume ratio. Another view is that a few specific interactions account for thermostability. This view forms the basis for mutational studies designed to alter an enzyme's thermostability (Argos et al., 1979; Menendez-Arias and Argos, 1989). Site-directed mutagenesis has been the method of choice in determining the contributions of specific groups or intermolecular interactions to the structure and intrinsic stability of a model protein, as done for example in the case of bacteriophage T4 lysozyme, ribonuclease HI or lactate dehydrogenase.
The main difficulties studying this subject are the only marginal stability of proteins and the high co-operativity and complexity of stabilizing interactions. The excess free energy of stabilization for thermostable proteins does not involve more than a few percent of the total number of weak interactions involved in the secondary, tertiary, and quaternary contacts within the molecule. To determine which of them is responsible for the differences in stability is not trivial. One solution to the problem would be the investigation of designed point mutants with known high-resolution structures and different thermal stabilities.
Keeping in mind that the increase in the free energy of stabilization,
![]()
![]() Gstab, for hyperthermophilic proteins is of the same
order of magnitude as the overall free energy of stabilization,
Gstab, for hyperthermophilic proteins is of the same
order of magnitude as the overall free energy of stabilization,
![]() GN
GN![]() U , it is evident that minute structural
alterations within a given protein molecule may suffice to cope with the
various extremes of temperature conditions. As a consequence, no general
strategy in terms of preferred amino acid exchanges or specific types of
intermolecular interactions is to be expected in going from mesophiles to
thermophiles.
U , it is evident that minute structural
alterations within a given protein molecule may suffice to cope with the
various extremes of temperature conditions. As a consequence, no general
strategy in terms of preferred amino acid exchanges or specific types of
intermolecular interactions is to be expected in going from mesophiles to
thermophiles.
Contents Hyperthermophiles and their proteins Factors involved in protein thermostability
FACTORS INVOLVED IN PROTEIN THERMOSTABILITY: HINTS FROM HYPERTHERMOPHILES
Much research has been devoted to determining which forces are responsible for thermostability. Contributions to stability have been elucidated by analyzing extremely stable proteins, such as proteins from hyperthermophilic microorganisms (Jaenicke, 1996).
Several factors have been shown to have an effect on the thermostability of a protein. Extrinsic factors non encoded in the sequence may be of importance in the increments of protein stability. For example, ions, cofactors, metabolites, or components covalently linked to the polypeptide chain may affect both protein stability and folding. Here, I am concerned only with intrinsic stabilization of proteins.
Mechanisms discussed in the context of increased intrinsic thermal stability of proteins are:
- the increase of the ion pairs content (Yip et al., 1995; Korndörfer et al., 195; Kelly et al., 1993; Korolev et al., 1995)
- the reduction of the number and volume of cavities (Russel et al., 1994)
- a tendency to form higher-order oligomers (Hecht et al., 1990)
- a decrease in flexibility at room temperature and in the length of surface loops, in particular those which connect secondary structure elements (Argos et al., 1979; Matthews, 1993; Russel et al., 1994)
- an optimization of electrostatic and hydrophobic interactions (Spassov et al., 1995)
- amino acids exchanges in order to increase internal hydrophobicity and helix propensity of residues in alpha-helices (Argos et al., 1979; Menendez-Arias and Argos, 1989)
- the replacement of amino acids sensitive to structure scrambling (Cys), deamidation (Asn, Gln), and oxidative damage (Met) (Michels et al., 1996).
In the following, the most important of them will be discussed.
- Thermal stability and amino acid composition: Thermal stability of proteins has been suggested on the basis of sequence comparisons to be due to many small structural differences. Comparing the sequences of ferrodoxins, glyceraldehyde-3-phosphate dehydrogenases, and lactate dehydrogenases from mesophiles and thermophiles and their corresponding three-dimensional structures, Argos et al. (1979) proposed rules for the temperature-dependent "gross traffic" of amino acids in going from mesophilic to thermophilic sequences. They suggest that relevant contributions to thermal stabilization may be attributed to both enhanced helicity (e.g., exchanges Ser to Ala, Val to Ala) and hydrophobicity (exchanges Gly to Ala, Ser to Ala, Ser to Thr/Gly/Ala, Asp to Asn, Glu), as well as the exchange Lys to Arg. This model has later been refined on the basis of data from a larger number (70) of protein sequences and structures then available (Menendez-Arias and Argos, 1989). Now a decreased flexibility and increased hydrophobicity, both preferably in alpha-helices regions, and to some extent in domain interfaces, were suggested as the main stabilizing principle. The ranking of the 5 most frequent amino acid exchanges from mesophilic to thermophilic enzymes was now seen to be Lys to Arg, Ser to Ala, Gly to Ala, Ser to Thr and Ile to Val.
The significance of most of these rules is ambiguous because the low extra free energy of thermal stabilization can be accumulated by innumerable combinations of subtle changes of local weak interactions. In all cases that have been studied so far, generalizations in terms of preferred amino acid exchanges have been unsuccessful. For instance, comparing exchanges from Homarus americanus (Ha) and Bacillus stearothermophilus (Bs) to Thermotoga maritima (Tm) GAPDHs (D-glyceraldehyde-3-phosphate dehydrogenase), Korndörfer et al. (1995) find that while some positions conform to published preferential exchanges, notable exceptions are observed and the predictive power of published statistics is limited. So, they suggest that the local structure of each mutated site must be carefully inspected to understand effects on the properties of proteins. For example, although substitutions by Gly may destabilize by an entropy increase of the unfolded state, they can also stabilize by release of steric hindrance. Bs and Tm GAPDHs are closely related in sequence and structure and should therefore be a good example to evaluate these models. Most frequent exchances from Ha (and Bs) to Tm GAPDHs are Gly to Ala, Ser to Thr and Ile to Val. These are numbers 3, 4 and 5 in the analysis containing all 70 sequences. The exchanges ranked 1 (Lys to Arg) and 2 (Ser to Ala) occur only twice and once, respectively, from Ha to Tm. Instead there are 4 exchanges Phe to Leu, which do not occur at all within the top ten exchanges. Although on the basis of Argos's rules it is expected that many of the frequent exchanges increase the Ala content of alpha-helices, Tm contains 7 Ala residues less than Ha and 14 less than Bs. For Ala residues found in alpha-helical regions of the compared structures, substitutions are mostly by residues classified as either indifferent or favorable, but no preference for a substitution with residues of higher helix propensity can be observed, nor is there a noticeable preference for alpha-helical positions in the changes towards Ala from "cold", Ha, to "hot", Tm. There is only one of the 5 Gly to Ala substitutions from Ha to Tm located in an alpha-helix. Also Jaenicke (1996), comparing Tm GAPDH to its mesophilic and thermophilic homologous, finds that the predictive power of traffic rules results in being limited. Analogous inconsistency has been observed in the case of PGK-TIM (Jaenicke et al., 1996). Nor are comparisons completely consistent for all protein families investigated. The content of phenylalanine residues has been reported to be increased in the thermotolerant members of the glyceraldehyde-3-phosphate dehydrogenase family (Zwickl et al., 1990), whereas it is decreased in thermostable members of the GluDH family (DiRuggiero et al., 1993).
In all cases that have been studied, the enzyme was shown to be isomorphous, that is, the structure was only locally perturbed. The results that have emerged illustrate how subtly the different weak interactions are balanced in native globular proteins and how much even marginal local strain in the three-dimensional structure may affect protein stability.
- Hydrophobic effect versus H-bonding: For 35 years, the prevailing view has been that the hydrophobic effect is the dominant force in protein folding. In his 1990 review, Dill concluded that "..there is now strong accumulated evidence that hydrophobicity is the dominant force of protein folding..". Hydrophobic interactions have been commonly interpreted in terms of entropic bonds, that is, by the increase in entropy caused by water release. However, high precision calorimetry has shown that there is a significant enthalpic contribution with may be ascribed to van der Waals forces. The importance of H-bonding was always clear, but whether it made a net favorable contribution to protein stability was not. In the 1995 edition of their textbook, Voet and Voet write: "..internal hydrogen bonding cannot significantly stabilize, and, indeed, may even slightly destabilize, the structure of a native protein relative to its unfolded state." Although a H-bond may contribute from 0.5 to 2 kcal/mol to the free energy of stabilization, their role in thermophilic proteins is seldom discussed. Presumably, this is because of their large numbers and the difficulty in determining which of them are important in any given structure. The more recent evidence, based on studies of mutant proteins, suggests that H-bonding and the hydrophobic effect make comparable contributions to the stability of globular proteins (Pace et al., 1996). Tanner et al. (1996) find that the stability of thermophilic GAPDHs derives more from electrostatic interactions (salt links and H-bonds) than hydrophobic interactions. They point out that thermostability of GAPDH from the extreme thermophile Thermus aquaticus correlates strongly with charged-neutral H-bonds. So they suggest that charged-neutral H-bonds may be more important than other hydrogen bonding pairs. There are two reasons that proteins may use charged-neutral H-bonds rather than salt links or neutral-neutral H-bonds to stabilize proteins. First, the desolvation penalty associated with burying a charged-neutral H-bond would be less than that of a salt link because only one charged residue is involved. Second, the enthalpic reward of a charged-neutral H-bond is greater than that of a neutral-neutral H-bond because of the charge-dipole interaction. This also indicates that the role of charged residues in stabilizing thermophilic proteins may not be limited to the formation of salt links.
- Salt links: The question of whether electrostatic interactions stabilize proteins is controversial. A significant increase in the number of salt-bridges has been reported for most structures of thermostable enzymes (Yip et al., 1995; Korndörfer et al., 195; Kelly et al., 1993; Korolev et al., 1995). However, site-directed mutagenesis experiments have not confirmed stabilizing contributions of ion pairs in thermostable proteins so far (Tomschy et al., 1994). Earlier structural comparisons between proteins from thermophilic and mesophilic organisms have shown that in thermostable proteins the number of surface ion pairs is increased (Perutz and Raidt, 1975). This finding has been further confirmed in recent structural comparisons of glyceraldehyde-3-phosphate dehydrogenase from Tm (Korndörfer et al., 1995), malate dehydrogenase from Thermus flavus (Kelly et al., 1993), DNA polymerase from Thermus aquaticus (Korolev et al., 1995) and citrate synthase from Thermoplasma acidophilum (Russel et al., 1994). At room temperature surface ion pairs destabilize the native protein, because of the incomplete coulombic compensation of solvation effects. However, at high temperature hydratation effects play a minor role that may result in a stabilizing net effect of ion pairs. In addition, the dielectric constant of water varies inversely with temperature due to the tendency of thermal motion to overcome the orientational effect.
Findings from studies on other oligomeric proteins show that the number of intersubunit ion pairs increases with thermostability (Perutz and Raidt, 1975). An increase of the number of ion pairs participating in subunit interactions has been also detected in a comparative study of the extremely thermotolerant GluDH from Pf (Yip et al., 1995) and the enzyme from the mesophile Clostridium symbiosum (Baker et al., 1992). On the other hand, Korndörfer et al. (1995) find a decrease of the number of intermolecular ion pairs from Bs GAPDH to the more thermotolerant Tm GAPDH. Tanner et al. (1996) find that in the case of Ta GAPDH the number of intrasubunit salt links correlates with thermostability, whereas the situation regarding intersubunit salt links was not as clear. This indicates that not all intersubunit ionic interactions are equally important for thermal stability of oligomeric enzymes.
- Compactness of the molecule: Increased compactness would lead to an increase of van der Waals interactions and higher protein stability. A possible measure of compactness is the comparison of accessible surface area and volume. The reduction of the solvent-accessible area and an increase of the fraction of buried hydrophobic atoms have been discussed as stabilizing principles for thermostable proteins (Chan et al., 1995; Spassov et al., 1995). In the case of GluDHs Knapp et al. (1997) find that the normalized accessible surface areas do not correlate with the thermal stability of the enzymes. This conclusion is supported by comparisons between different glyceraldehyde-3-phosphate dehydrogenases (Korndörfer et al., 1995) and indole-3-glycerolphosphate synthases (Hennig et al., 1995). However, if the contributions of nonpolar, polar and charged amino acids to the accessible surface areas are analysed separately, the thermostable GluDHs expose less hydrophobic and more charged residues.
The volume of the cavities in the subunits decreases significantly with increasing thermal stability of the enzymes. In GluDH from Pf a significant increase in Ile residues has been detected (Britton et al., 1995). Ile residues are able to adopt several rotamer conformations and are therefore extremely suitable to fill various voids in the protein core.
It has been suggested that decreased surface to volume ratio is related to thermostability. Tanner et al. (1996) find that the surface to volume ratio of the Thermophiles are about 12-22% smaller than that of the psychrophile. Decreases in the surface to volume ratio in their study result from both an increase in the volume enclosed by the molecular surface and a decrease in the area of the molecular surface. One way for a three-dimensional object to decrease its surface area and to increase its volume is to become more spherical. A spherical protein would minimize the surface area and maximize the number of residues in the interior of the protein. Maximization of the cohesive forces inside the protein is driving the tendency toward a more spherical protein. Therefore, buried salt links, buried H-bonds, and hydrophobic interactions are more likely to confer thermostability than are interactions on the surface such as solvent exposed salt links. But this is inconsistent with the idea that thermal stabilization of proteins can be provided by salt-bridges on the molecular surface rather than buried ones.
- Structural elements: : Maximum thermostability has been observed in all-alpha, all-beta, and alpha beta proteins (e.g., rop, beta gamma crystallins, immunoglobulins, and oligomers such as NAD-dependent dehydrogenases) (Jaenicke et al., 1996).
Contents Protein thermostability Examples
A FEW EXAMPLES
In the following, representative examples will be discussed in order to illustrate some characteristics of hyperthermophilic proteins and to provide perspectives with respect to possible generalizations in terms of factors involved in protein thermostability.
The first three examples allow to understand the role that ion pairs, H-bonds and hydrophobic interactions can play in protein thermostability.
- GluDH (Glutamate dehydrogenase): Knapp et al. (1997) have chosen GluDH from Tm to study the mechanisms that may lead to the extreme thermal stability of this enzyme because glutamate dehydrogenases are well studied and structural information is already available for an enzyme from a mesophilic bacterium (Clostridium symbiosum) as well as a high thermostable enzyme (Pyrococcus furiosus). GluDHs form hexamers arranged in 32 symmetry. The enzyme plays an important key role in all organisms: it links the carbon and nitrogen metabolism by catalyzing the oxidative deamination of L-glutamate to form 2-oxoglutarate and ammonia using the cofactors NAD(P)H.
Click here for a PDB file and a gif image of GluDH from Clostridium symbiosum.
Click here for a PDB file and a gif image of GluDH from Pyrococcus furiosus.
The structural comparison of the three enzymes suggests that an increased number of ion pairs and the extent of ion pair networks modulate their thermal stability. The comparison indicates as well that increased hydrophobic interactions in the protein core, and in the case of Tm at the subunit interface, may contribute to the stability of thermostable GluDHs.
The structure-based alignment of the primary structures suggests that an increase in thermal stability may be achieved by 1) an increase in rigidity by a reduction of Gly residues; 2) a decrease in the sulphur content; 3) an increase of hydrophobic contacts in Pf GluDH by Val to Ile mutations; and 4) a modulation of the subunit flexibility: most of the sequence variations between GluDHs have been found in the hinge region connecting both domains. It is probable that sequence variations in the hinge regions influence the domain movements within the subunit.
ProtParam tool on the ExPaSy server allows you to analyse primary sequence parameters. Compare the amino acid composition of the three GluDHs and the aliphatic index at the following links (Cs, Pf, Tm). The aliphatic index is defined as the relative volume of a protein occupied by aliphatic side chains (Ala, Val, Ile and Leu). It may be regarded as a positive factor for the increase of thermostability of globular proteins. Its value is 92.52 for Tm, 88.24 for Pf, 79.47 for Cs.
- GAPDHs (Glyceraldehyde-3-phosphate dehydrogenases). The availability of sequence and structural information on GAPDH enzymes from a wide variety of organisms makes this enzyme ideal for investigating mechanisms of protein stability at high temperature.
Korndörfer et al. (1995) solved the crystal structure of holo-glyceraldehyde-3-phosphate dehydrogenase from the hyperthermophilic bacterium Thermotoga maritima at 2.5 Å resolution ( click here for a PDB file) and compared it to the models of holo-GAPDH from Bs (Bacillus stearothermophilus) and Ha (Homarus americanus). The final model of Tm GAPDH is made up of 2 monomers in the asymmetric unit with 332 amino acid residues each. These monomers are related by approximate 2-fold symmetry and a tetramer is built up by 222 crystallographic symmetry. This enzyme catalyzes the oxidative phosphorylation of the substrate glyceraldehyde-3-phosphate to D-1,3-bisphosphoglycerate, with NAD+/NADH as coenzyme.
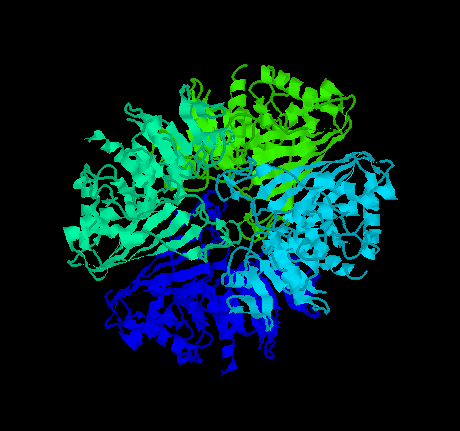 Click here
for a gif image of the tetramer of Tm GPHD
Click here
for a gif image of the tetramer of Tm GPHD
The structure is higly homologous with the enzyme from both other thermophilic and mesophilic bacteria. Taking all known structures together, about 33% of the residues are identical. Comparing the sequences of the enzyme from Tm, Bs and Ta (Thermus aquaticus), 63 and 59% identity are observed; only 8% of the exchange are nonconservative. This means that the 3 homologous GAPDH enzymes, with denaturation temperatures of 110, 80, and 68°C, differ in about one-third of their amino acid sequence, that is, in 100 of 330 residues. Which of the different residues are responsible for the change in thermal stability is not easy to find.
Salt-bridges play an important role in the stabilization of GAPDHs. Many of the buried intermolecular ion pairs are part of a cluster of ionic bonds in direct or indirect contact with Arg10, which is conserved in all known (hyper-) thermophilic GAPDHs. The conserved salt bridge between Lys108 and Glu94 is located in the core of the binding domain and is probably crucial for maintenance of the three-dimensional structure of the site of NAD+ coordination. Lys306 and Asp241 are located in the interface between the catalytic domains of two subunits and therefore shielded from the solvent by the neighbouring subunit. The conserved salt bridge on the S-loop between Asp186 and Arg197, too, is only shielded from the solvent by neighbouring subunits. The latter four residues are highly conserved in over 20 GAPDH sequences. Destruction of these interactions could infer consequences on the interactions of subunits within the tetramer. A large number of extra salt-bridges have been detected in Tm and are considered to be an important factor contributing to the high thermostability of this protein. The case of Tm GAPDH confirms the hypothesis that thermal stabilization of proteins can be provided by salt bridges on the molecular surface rather than in the inner core of the molecule. All additional ion pairs attributable to the thermostability of Tm GAPDH are located in the periphery of the tetramer (see table 1).
| Residue 1 | Residue 2 | Ha | Bs | Tm |
| Arg10 | Glu314 | ++++ | ++++ | ++++ |
| Arg10 | Asp47 | ++++ | ++++ | ++++ |
| Arg20 | Asp323 | - | - | ++++ |
| Arg20 | Asp326 | - | - | ++++ |
| Lys56 | Glu58 | - | - | ++ |
| Lys81 | Glu76 | - | - | ++(++) |
| Lys81 | Asp78 | - | - | ++++ |
| Lys101 | Glu103 | - | +(+++) | - |
| Arg102 | Glu106 | - | - | ++++ |
| Arg102 | Asp125 | - | ++++ | ++++ |
| Lys101 | Glu103 | - | - | ++(++) |
| Lys107 | Asp78 | - | - | ++(++) |
| Lys107 | Asp104 | - | +++(+) | - |
| Lys114 | Asp90 | - | ++(++) | ++(++) |
| Lys136 | Asp135 | - | +(+) | - |
| Lys159 | Glu163 | - | ++++ | ++(++) |
| Hys190 | Asp181 | - | ++++ | ++++ |
| Arg195 | Asp181 | - | ++++ | ++++ |
| Arg195 | Asp192 | - | ++++ | (++++) |
| Arg197 | Asp186 | +++(+) | ++++ | ++++ |
| Arg245 | Glu169 | ++++ | - | - |
| Lys256 | Glu275 | +(++) | - | - |
| Lys260 | Glu275 | +(+++) | (+++) | (+) |
| Lys260 | Glu264 | +(+++) | ++++ | ++++ |
| Arg266 | Glu163 | - | - | ++++ |
| Arg288 | Asp286 | ++++ | - | - |
| Lys303 | Glu249 | - | (++) | (++) |
| Total | 55(17) | 59(33) | 73(32) |
Residue numbering and residue types have been taken from Tm GAPDH, except where no salt bridge at all is found in Tm GAPDH. For the corresponding residue numbers and types for Ha and Bs, see the Brookhaven Protein Data Bank. Only ion pairs up to a distance of 4.0 Å are included. + stands for an ion pair in each of the four chains of the tetramer. (+) stands for an ion pair within up to 6.0 Å that failed the 4.0 Å distance criterion but has corresponding pairs in either another subunit or one of the other enzymes shown here. (Taken from Jaenicke et al., 1996, p.219).
Another factor contributing to the stability of Tm GAPDHase may be the compactness of the molecule, which results in an increase of van der Waals interactions. A possible measure for compactness is the accessible surface area. For the three structures compared (Ha, Bs, Tm), the surface area of hydrophobic residues buried in tetramer contacts increases with thermostability (Korndörfer et al., 1995). This may lead to a stabilization of the tetramer with respect to subunit dissociation.
In their excellent work, Tanner et al. (1996), after solving the crystal structure of holo D-glyceraldehyde-3-phosphate dehydrogenase from the extreme thermophilic Thermus aquaticus (optimal growth temperature: 70-75°C) studied the determinants of its thermostability comparing this structure to four other GAPDHs from holo Homarus americanus (optimal growth temperature: 20°C), apo Homarus americanus (optimal growth temperature: 20°C), holo Bacillus stearothermophilus (optimal growth temperature: 58°C), Thermotoga maritima (optimal growth temperature: 80°C).
Click here for PDB files of GPDH from Thermus aquaticus, holo Homarus americanus, apo Homarus americanus, holo Bacillus stearothermophilus, Thermotoga maritima.
They find a strong correlation between thermostability and the number of H-bonds between charged side chains and neutral partners. These charged-neutral hydrogen bonds provide electrostatic stabilization without the heavy desolvation penalty of salt links. The stability of thermophilic GAPDHs is also correlated with the number of intrasubunit salt links and total hydrogen bonds. Charged residues, therefore, play a dual role in stabilization by partecipating not only in salt links but also in hydrogen bonds with a neutral partner. Hydrophobic effects allow for discrimination between thermophiles and psychrophiles, but not within the GAPDH thermophiles. There is, however, an association between thermostability and decreasing enzyme surface to volume ratio. A four-residue salt link network, a hydrogen bond near the active site (see a gif image), an intersubunit salt link, and several buried Ile residues are conserved in Ta and Tm, and not in the less thermostable GAPDHs: this is consistent with the view that thermostability is correlated with the overall number of salt links and H-bonds.
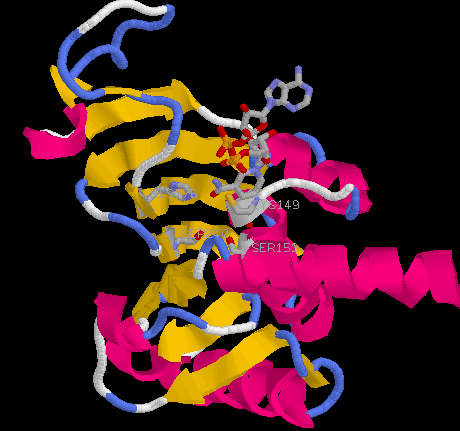 Click here for a gif image
of Ser153-Ser240 additional H-bond in the active
site region of the R subunit of Ta GAPDH. The active site residues
Cys149 and His176 and the coenzyme NAD+ are also shown. The distance between
the OG atoms of the two Ser residues is 3.3 Å. This H-bond provides
some stabilization of the active site region, linking the helix where
Ser153 is located to a beta strand. Residues 153 and 240 are Ser and Thr
in Tm and Tm structure also shows these residues within hydrogen
bonding distance. This H-bond is not present in the Ha and Bs
structures because in their sequences residues 153 and 240 are both
replaced by Cys and Val.
Click here for a gif image
of Ser153-Ser240 additional H-bond in the active
site region of the R subunit of Ta GAPDH. The active site residues
Cys149 and His176 and the coenzyme NAD+ are also shown. The distance between
the OG atoms of the two Ser residues is 3.3 Å. This H-bond provides
some stabilization of the active site region, linking the helix where
Ser153 is located to a beta strand. Residues 153 and 240 are Ser and Thr
in Tm and Tm structure also shows these residues within hydrogen
bonding distance. This H-bond is not present in the Ha and Bs
structures because in their sequences residues 153 and 240 are both
replaced by Cys and Val.
Adams and Kletzin (1996) have studied Pyrococcus woesei GAPDH (optimal growth temperature: 100°C). The amino acid sequence of the Pyrococcus woesei enzyme was about 50% identical with those of mesophilic and thermophilic GAPDH enzymes obtained from both archaea and bacteria. Sequence comparisons indicated that the hyperthermophilic enzyme had an increase in average hydrophobicity and a decrease in average chain flexibility compared to the less thermophilic proteins. Thermal denaturation of the enzyme was shown to involve, at least in part, deamidation of asparagine residues.
Construction of hybrid enzymes between the GAPDH from the mesophilic Methanobacterium bryantii (optimal growth temperature: 37°C) and the thermophilic Methanothermus fervidus (optimal growth temperature: 83°C) by recombinant DNA tecniques (Biro et al., 1990) revealed that a short C-terminal fragment of the Methanothermus fervidus enzyme contributes largely to its thermostability. This C-terminal region appears to be homologous to the alpha3-helix of eubacterial and eukaryotic GAPDHs which is involved in the contacts between the two domains of the enzyme subunit. Exclusively these contacts clamp the C-terminal, so-called catalytic domain and the N-terminal domain together. Site-directed mutagenesis experiments indicate that hydrophobic interactions play an important role in these contacts. To define the interactions by which the short C-terminal fragment (20-25 residues) stabilizes the native conformation of the GAPDH from Methanothermus fervidus, single residues in that region were exchanged and the effect of the replacement on thermostability was tested. For the exchange the authors focused on position 323 where the respective residues in the different GAPDHs show an increase in hydrophobicity from mesophilic to thermophilic structures: at that position the enzymes from the closely related mesophilic methanogens Methanobacterium bryantii and Methanobacterium formicicum possess Ser, the more thermostable enzyme from Methanothermus fervidus Tyr and the enzyme from Pyrococcus woesei with the highest thermostability Trp. The change to Ser (mutant GAPDH Y323S) resulted in a decrease of thermostability by 4.5°C, the change to Trp (mutant GAPDH Y323W) in an increase of thermostability by 1.3°C as compared to the wild-type enzyme. The influence of the performed exchanges on thermostability can be explained by the involvement of the residue at position 323 in interdomain contacts governing the rigidity and thus the stability of the protein conformation.
- Alpha-Amylases: Although most alpha-amylases derived from animals, plants as well as microorganisms keep their unique properties only in the normal physiological conditions, there are some microbial alpha-amylases which exhibit exceptional stability form thermophiles, acidophiles, alkalophiles, and halophiles. Alpha-amylase is a member of (alpha/beta)8 barrel enzyme family, this motif (a barrel domain of 8 parallel beta-strands surrounded by 8 alpha-helices) was first observed in triose phosphate isomerase. The alpha-amylases with known three-dimensional structure are multidomain single polypeptide-chain proteins. Besides the (alpha/beta)8 barrel they all comprise an antiparallel beta-stranded domain. Alpha-amylases are enzyme capable of hydrolyzing starch. Active site is localized in the cleft of the (alpha/beta)8 barrel domain. All alpha-amylases are metalloenzymes containing at least one Ca2+ ion per enzyme molecule, which is essential for activity and stability.
The extra thermostability of thermophilic Bacillus licheniformis alpha-amylase (click here for a gif image and a pdb file) has been found (Janecek and Balaz, 1992) to be mainly due to additional salt bridges involving a few specific lysine residues. Similarly the Bacillus stearothermophilus alpha-amylase as well as a mutant alpha-amylase from Bacillus amyloliquefaciens have been suggested to be stabilized against thermal denaturation through ionic interaction.
The last two examples refer to the suggestion that an higher state of association can contribute to thermal stability.
- LDH (Lactate dehydrogenase) from Tm (link to a PDB file): The enzyme is highly homologous to LDHs from other sources, including thermophilic bacteria. One of the effects assumed to contribute to its anomalous thermostability is the tendency of the tetrameric enzyme to form octamers (Jaenicke, 1996). Fragmentation studies have clearly shown that there is a continuous decrease in stability in going from the tetramer to the dimer, monomer, and, finally, to domains (Opitz et al., 1987).
- Enolase from Tm: Enolase is a Mg2+-dependent homooctamer with exceedingly high intrinsic thermostability (temperaturemax = 94°C) and specific activity (2000 U/mg at optimal temperature; corresponding values for the mesophilic homologs are 70 U/mg for the enzyme from B. megaterium, 160 for E. coli, and 450-900 for Thermus aquaticus). Here is a case where a protein, during evolution, succeeded in optimizing more than one function, exhibiting both maximum catalytic efficiency and high stability, even beyond the growth temperature of Tm. As Jaenicke (1996) highlights, the enzyme commonly forms dimers, so that the higher state of association has been considered a possible mechanism of thermal adaptation. Because octameric enolases have also been found in nonthermophilic bacteria, this hypothesis cannot be correct. Moreover, enolase from the hyperthermophilic archaeon Pyrococcus furiosus has been reported to be a dimer. Thus there is no clear correlation of the stability of Tm enolase to its state of association.
Contents Factors involved in protein thermostability Conclusions
Whilst major advances have been made in recent years thanks to the findings from the new field of hyperthermophiles, our present knowledge of factors involved in protein stability is still relatively limited and increased research effort will be required.
As Jaenicke (1996) highlights, no general and
quantitative correlations between the increase in stability and alterations
in the three-dimensional structure of proteins can be given at this time,
for the following reasons: 1) only marginal increments to the free energy
of stabilization,
![]()
![]() Gstab,, are required to extend the range of thermal
stability within the biologically relevant temperature range; 2) structural
differences between mutants commonly are not strictly focused at the exchanged
residues, but spread over large parts of the molecule.
Gstab,, are required to extend the range of thermal
stability within the biologically relevant temperature range; 2) structural
differences between mutants commonly are not strictly focused at the exchanged
residues, but spread over large parts of the molecule.
It is also necessary to pay attention to bias and inconsistency of findings from different researches.
In comparing crystal structures solved at different times in different laboratories, one concern is that the reported differences in numbers of electrostatic interactions are due to variations in refinement software and strategy. For example, some refinement programs use force fields (see the PPS course) that include not only bond, angle, and dihedral terms, but also nonbonded electrostatic terms based on partial charges. The degree to which the observed salt links and hydrogen bonds are biased by the force field parameters, data collection, and processing and refinement is not known. These concerns are expected to be minimized at higher resolution, suggesting a need for new higher resolution structure of both thermophilic and mesophilic enzymes.
Bearing in mind that a limited number of subtle changes in local weak interactions is sufficient to shift the stability profile from the mesophilic to the thermophilic temperature range, x-ray crystallography at a resolution of better than 2 Å will be also required in order to eventually explain the thermal stability of the enzymes.
In the light of the inconsistency among the results of different researches as regards mechanisms of protein stabilization, comparative studies must be extended to a larger body of enzymes from thermophilic and mesophilic organisms to allow differentiation between species-specific variations and those modifications necessary for the thermal stability of the enzyme under consideration.
Additional experiments like site-directed mutagenesis together with thermodinamic stability measurements are necessary to demonstrate the effect of the variations detected by the structural comparison of the thermotolerance of proteins.
Evolution result in biomolecules with optimum function rather than maximum stability, in order to optimize a multifunctional system as are proteins (folding, assembly, ligand binding, catalysis, turnover). In cases such as Tm enolase, both requirements coincide. The molecular basis of the stability of proteins like this is still not clear and further research in this subject will contribute to deepen our knowledge of how nature is able to balance so different requests such as flexibility and stability.
Adams, M. W. W. and Kletzin, A. (1996). Oxidoreductase-type Enzymes and Redox Proteins involved in Fermentative Metabolisms of Hyperthermophilic Archaea. Advances in Protein Chemistry, 48, 101-179.
Argos, P., Rossmann, M. G., Grau, U., Zuber, H., Frank, G., and Tratschin, J. D. (1979). Thermal stability and Protein Structure. Biochemistry, 18, 5698-5703.
Baker, P. J., Britton, K. L., Engel, P. C., Farrants, G. W., Lilley, K. S., Rice, D. W. and Stillman, T. J. (1992). Subunit Assembly and Active Site Location in the Structure of Glutamate Dehydrogenase. Proteins: Struct. Funct. Genet., 12, 75-86.
Baross, J. A. and Holden, J. F. (1996). Overview of Hyperthermophiles and their Heat-shock Proteins. Advances in Protein Chemistry, 48, 1-34.
Biro, J., Fabry, S., Dietmaier, W., Bogedain, C. and Hensel, R. (1990). Engineering Thermostability in Archaebacterial Glyceraldehyde-3-phosphate Dehydrogenase. Hints for the important role of Interdomain Contacts in Stabilizing Protein Conformation. FEBS LETTERS, 275, 1, 2, 130-134.
Britton, K. L., Baker, P. J., Borges, K. M. M., Engel, P. C., Pasquo, A., Rice, D. W., Robb, F. T., Scandurra, R., Stillman, T. J. and Yip, K. S. P. (1995). Insights into Thermal Stability from a Comparison of the Glutamate Dehydrogenase from Pyrococcus furiosus and Thermococcus litoralis. Eur. J. Biochem., 229, 688-695.
Chan, M. K., Muklund, S., Kletzin, A., Adams, M. W. W. and Rees, D. C. (1995). Structure of a Hyperthermophilic Tungstoperin Enzyme, Aldheyde Ferredoxin Oxidoreductase. Science, 267, 1463-1469.
DiRuggiero, J., Robb, F. T., Jagus, R., Klump, H. H., Borges, K. M. M., Kessel, M., Mai, X. and Adams, M. W. W. (1993). Characterization, Cloning and In vitro Expression of the Extremely Thermostable Glutamate Dehydrogenase from the Archaeon, ES4. J. Biol. Chem., 268, 17767-17774.
Dill, K. (1990). Dominant Forces in Protein Folding. Biochemistry, 29, 7133-7155.
Hecht, K., Wrba, A. and Jaenike, R. (1990). Catalytic Properties of Thermophilic LDH and Halophilic MDH at High Temperature and Low Water Activity. Eur. J. Biochem., 183, 69-74.
Hennig, M., Darimont, B., Sterner, R., Kirschner, K. and Jansonius, J. N. (1995). 2.0 Å Structure of Indole-3-Glycerol Phosphate Synthase from the Hyperthermophile Sulfolobus solfataricus: Possible Determinants of Protein Stability. Structure, 3, 1295-1306.
Herbert, R. A. (1992). A Perspective on the Biotechnological Potential of Extremophiles. Tibtech, 10, 395-402.
Jaenicke, R. (1996). Stability and Folding of Ultrastable Proteins: Eye Lens Crystallins and Enzymes from Thermophiles. FASEB J., 10, 84-92
Jaenicke, R., Schurig, H., Beaucamp, N. and Ostendorp, R. (1996). Structure and Stability of Hyperstable Proteins: Glycolytic Enzymes from Hyperthermophilic Bacterium Thermotoga Maritima. Advances in Protein Chemistry, 48, 181-269.
Janecek, Æ. and Baláz, Æ. (1992). Alpha-Amylases and Approach Leading to their Enhanced Stability. FEBS LETTERS, 304, 1, 1-3.
Kelly, C. A., Nishiyama, M., Ohnishi Y., Beppu, T. and Birktoft, J. J. (1993). Determinants of Protein Stability in the 1.9-Å Crystal Structure of Malate Dehydrogenase from the Thermophilic Bacterium Thermus flavus. Biochemistry, 32, 3913-3922.
Knapp, S., M. de Vos, W., Rice, D. and Ladenstein, R. (1997). Crystal Structure of Glutamate Dehydrogenase from the Hyperthermophilic Eubacterium Thermotoga maritima at 3.0 Å Resolution. J. Mol. Biol., 267, 916-932.
Korndörfer, I., Steipe, B., Huber, R., Tomschy, A. and Jaenicke, R. (1995). The Crystal Structure of Holo-glyceraldehyde-3-phosphate Dehydrogenase from the Hyperthermophilic Bacterium Thermotoga maritima at 2.5 Å Resolution. J. Mol. Biol., 246, 511-521.
Korolev, S., Nayal, M., Barnes, W. M., Di Cera, E. and Waksman, G. (1995). Crystal Structure of the Large Fragment of Thermus aquaticus DNA Polymerase I at 2.5 Å Resolution: Structural Basis for Thermostability. Proc. Natl Acad. Sci. Usa, 92, 9264-9268.
Madigan, M. T. and Marrs, B. L. (1997). Gli Estremofili. Le Scienze, 346, 78-85.
Matthews, B. W. (1993). Structural and Genetic Analysis of Protein Stability. Annu. Rev. Biochem., 62, 139-160.
Menendez-Arias, L. and Argos, P. (1989). Engineering Protein Thermal Stability. Sequence Statistics Point to Residue Substitutions in alpha-Helices. J. Mol. Biol., 206, 397-406.
Michels, P. C., Hei, D. and Clark, D. S. (1996). Pressure Effects on Enzyme Activity and Stability at High Temperatures. Advances in Protein Chemistry, 48, pp. 341-375.
Opitz, U., Rudolph, R., Jaenicke, R., Ericsson, L. and Neurath, H. (1987). Biochemistry. 26, 1399-1406.
Pace, C. N., Shirley, B. A., Mcnutt M. and Gajiwala K. (1996). Forces Contributing to the Conformational Stability of Proteins. FASEB J., 10, 75-83.
Perutz, M. F. and Raidt, H. (1975). Stereochemical Basis of Heat Stability in Bacterial Ferredoxins and in Haemoglobin A2. Nature (London), 255, 256-259.
Russel, R. J. M., Hough, D. W., Danson, M. J. and Garry, L. T. (1994). The Crystal Structure of Citrate Synthase from the Thermophilic Archaeon Thermoplasma acidophilum. Structure, 2, 1157-1167.
Schäfer, G. (1992). Extremophiles: Fascinating Organisms with Surprising Capabilities. Journal of Bioenergetics and Biomembranes, 24, 6, 525-527.
Spassov, V. Z., Karshikoff, A. D. and Ladenstein, R. (1995). The Optimization of Protein-solvent Interactions: Thermostability and the Role of Hydrophobic Interactions. Protein Sci., 4, 1516-1527.
Tanner, J. J., Hecht, R. M. and Krause, K. L. (1996). Determinants of Enzyme Thermostability Observed in the Molecular Structure of Thermus aquaticus D-Glyceraldehyde-3-phosphate Dehydrogenase at 2.5 Å Resolution. Biochemistry, 35, 2597-2609.
Tomschy, A., Böhm, G. and Jaenicke, R. (1994). The Effect of Ionpairs on the Thermal Stability of D-Glyceraldehyde-3-phosphate Dehydrogenase from the Hyperthermophilic Bacterium Thermotoga maritima. Prot. Eng., 7, 1471-1478.
Voet, D. and Voet, J. G. (1995). Biochemistry, J. Wiley & Sons, Inc.
Wrba, A., Schweiger, A., Schultes, V. and Jaenicke, R. (1990). Extremely Thermostable D-Glyceraldehyde-3-phosphate Dehydrogenase from the Eubacterium Thermotoga maritima. Biochemistry, 29, 7584-7592.
Yip, K. S. P., Stillman, T. J., Britton, K. L., Artymiuk, P. J., Baker, P. J., Sedelnikova, S. E., Engel, P. C., Pasquo, A., Chiaraluce, R., Consalvi, V., Scandurra, R. and Rice, D. W. (1995). The Structure of Pyrococcus furiosus Glutamate Dehydrogenase Reveals a Key Role for Ionpair Networks in Maitaining Enzyme Stability at Extreme Temperatures. Structure, 3, 1147-1158.
Yip, K. S. P., Baker, P. J., Britton, K. L., Engel, P. C., Rice, Sedelnikova, S. E., Stillman, T. J., Pasquo, A., Chiaraluce, R., Consalvi, V. and Scandurra, R. (1995). Crystallization of the NAD(P) Dependent Glutamate Dehydrogenase from the Hyperthermophilic Pyrococcus furiosus. Acta crystallog. sect. D., 51, 249-242.
Zwickl, P., Fabry, S., Bogedain, C., Haas, A. and Hensel, R. (1990). Glyceraldehyde-3-phosphate Dehydrogenase from the Hyperthermophilic Archaebacterium Pyrococcus woesei: Characterization of the Enzyme, Cloning and Sequencing of the Gene, and Expression in Escherichia Coli. J. Bacteriol., 172, 4329-4338.
{kind=link}
{kind=link}
{kind=link}