This page under construction!
This page under construction!
Determination of the bR's Structure
We have already seen that bR is a bundle of seven transmembrane helices. This structural feature can also be found in other integral membrane receptor proteins, such as those of the adrenergic, cholinergic, serotonin, somatostatin, and other receptor families. Rao et. al. have proposed that "...the packing of seven helices together may represent a uniqely stable arrangement that has been achieved through a process of convergent evolution." The current model of bR's structure, is based on Henderson's et. al. work using electron cryo-microscopy, and a refinement proposed by Chou et. al. using an energy-based approach. Here I will attempt to examine their work.
Model for the Structure of Bacteriorhodopsin Based on High-Resolution Electron Cryo-Microscopy
(reference)
Prologue: A brief account of indirect work leading to several conclusions concerning the structure of bR
Much data for the structure of bR was obtained by means other than obtaining data from diffraction patterns. Here is a very brief outline of such experiments. It is beyond the scope of these pages to deal with this type of work in the detail that it deserves.
Khorana and colleagues have investigated a large number of single amino-acid mutants of bR. They have shown that mutations to Asp85, Asp96 and, to a lesser extent, Asp212 have effects on proton pumping. This type of experiment, combined with data from other experiments (such as linear dichroism measurements which helped determine the orientation of the retinal absorption dipole), helped in the definition of "anchoring points" concerning the location of amino-acid residues in bR, simply by determining their effect on the functionality of bR. Another method used was comparison with related proteins within the family, such as halorhodopsin, to determine amino-acid homology. The degree of homology between the two proteins was greater among amino-acids which were expected to be directed inwards, towards the retinal binding site.
The absorption spectrum of bR is disturbed by changes to other aa residues: Trp86, Trp137, Trp182, Trp189,
Arg 82, Thr89, and Asp115.
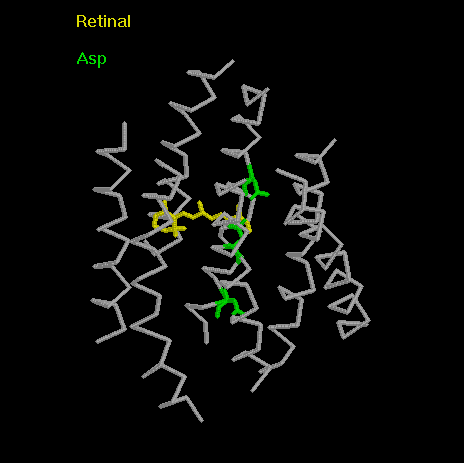
View of bR with Asp85, Asp96, Asp212
marked.
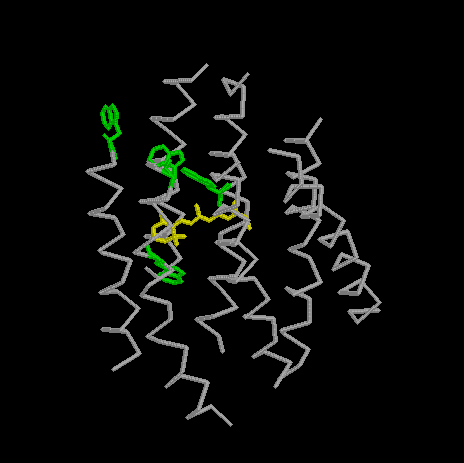
View bR with Trp86, Trp137, Trp182, and Trp189 marked.
View bR withThr89, and Asp115 marked.
In general, this data provides evidence concerning the orientations of the alpha-helices in the structure, showing
which side-chains point inwards towards the retinal binding site, and the proton channel. Generally, the data
is in agreement with the idea that hydrophyllic faces point inwards, towards the active site
(the channel, evidently, contains water), and the hydrophobic faces ouwards, towards the lipid bilayer.
As stated in the introduction, Henderson & Unwin's initial work from 1975 yielded a structure with a 7-A horizontal resolution. The structure was refined in 1990 using electron cryo-microscopy. Diffraction patterns obtained by that method enabled structure determination up to a 3.5-A vertical resolution, and a 10-A horizontal one.
The problem of resolution, despite improvements made in electron-diffraction techniques, still remains. The main density in the 3D density map is that of the alpha-helices. In addition, well-resolved features on the three-dimensional density map include side chains of bulky aromatic amino-acids such as Phe, Tyr, Trp. A very dense peak in the map is obtained from the beta-ionone ring of retinal.
Using these features as anchor-points, plus the data obtained from experimental studies (e.g. site-directed mutagenesis), an atomic model between residues 8 and 225 was constructed. A mechanism for proton-pumping was also suggested.
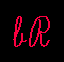
Secondary structure model of bR based on electron cryo-microscopy results. Helical segments are outlined by a box. Residues within a red circle lie within the proton channel. The residues in a black circle are those for which heavy-atom coordinates have not yet been determined. Krt+ designates the retinal-Lys216 all-trans isomer.
A comprehensive Kinemage created by D. Donnelly et. al. is provided here. It is based on PDB entry 1BRD, the all-trans conformation of bR as determined by Henderson et. al.
An Energy Based approach to packing the 7-helix bundle of bacteriorhodopsin
An additional refinement, using an energy based approach to the packing of the 7-helix bundle was proposed by Chou et. al. in 1992.
Authors' conclusions:
{kind=link}
{kind=link}
{kind=link}