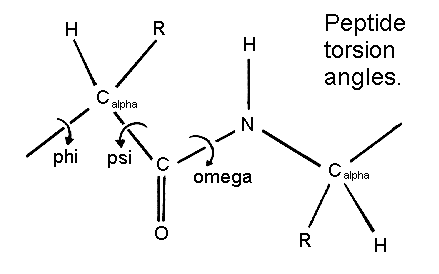
The figure below shows the three main chain torsion angles of a polypeptide. These are phi, psi and omega.
The planarity of the peptide bond restricts omega to 180 degrees in very nearly all of the main chain peptide bonds. In rare cases omega = 0 degrees for a cis peptide bond which, as stated above, usually involves proline.
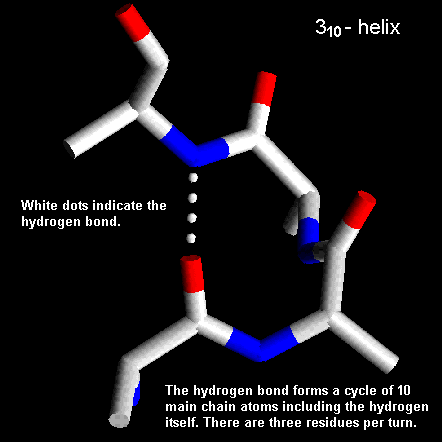
Pauling and Corey twisted models of polypeptides around to find ways of getting the backbone into regular conformations which would agree with alpha-keratin fibre diffraction data. The most simple and elegant arrangement is a right-handed spiral conformation known as the 'alpha-helix'.
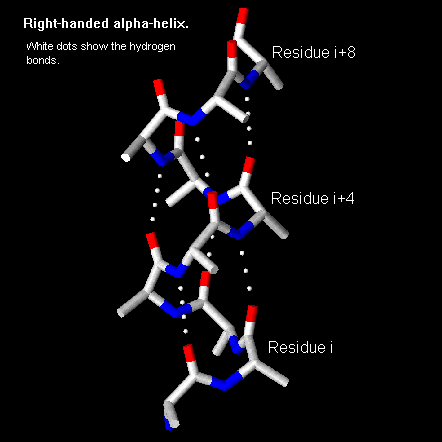
The figure below shows how a right-handed helix differs from a left-handed one. An easy way to remember this is to hold both your hands in front of you with your thumbs pointing up and your fingers curled towards you. For each hand the thumbs indicate the direction of translation and the fingers indicate the direction of rotation.
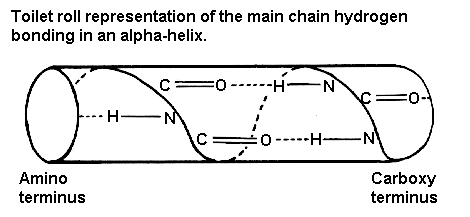
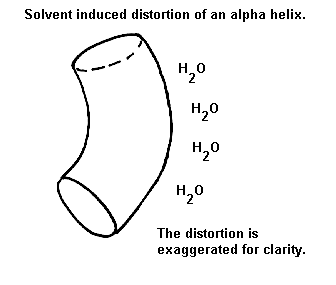
The majority of alpha-helices in globular proteins are curved or distorted somewhat compared with the standard Pauling-Corey model. These distortions arise from several factors including:
Pauling and Corey derived a model for the conformation of fibrous proteins known as beta-keratins. In this conformation the polypeptide does not form a coil. Instead, it zig-zags in a more extended conformation than the alpha-helix. Amino acid residues in the beta-conformation have negative phi angles and the psi angles are positive. Typical values are phi = -140 degrees and psi = 130 degrees. In contrast, alpha-helical residues have both phi and psi negative. A section of polypeptide with residues in the beta-conformation is refered to as a beta-strand and these strands can associate by main chain hydrogen bonding interactions to form a beta sheet.
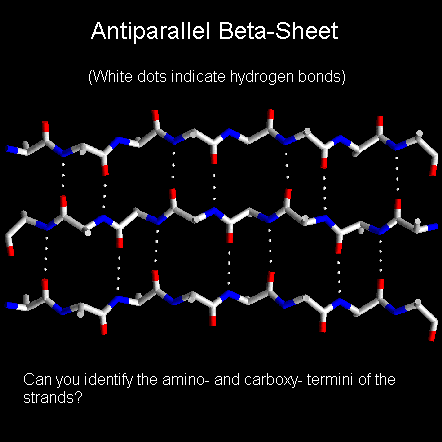
In a beta-sheet two or more polypeptide chains run alongside each other and are linked in a regular manner by hydrogen bonds between the main chain C=O and N-H groups. Therefore all hydrogen bonds in a beta-sheet are between different segments of polypeptide. This contrasts with the alpha-helix where all hydrogen bonds involve the same element of secondary structure. The R-groups (side chains) of neighbouring residues in a beta-strand point in opposite directions.
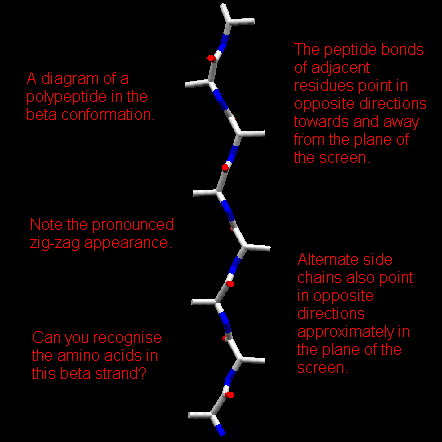
Imagining two strands parallel to this, one above the plane of the screen and one behind, it is possible to grasp how the pleated appearance of the beta-sheet arises. Note that peptide groups of adjacent residues point in opposite directions whereas with alpha-helices the peptide bonds all point one way.
The axial distance between adjacent residues is 3.5 Angstroms. There are two residues per repeat unit which gives the beta-strand a 7 Angstrom pitch. This compares with the alpha-helix where the axial distance between adjacent residues is only 1.5 Angstroms. Clearly, polypeptides in the beta-conformation are far more extended than those in the alpha-helical conformation.
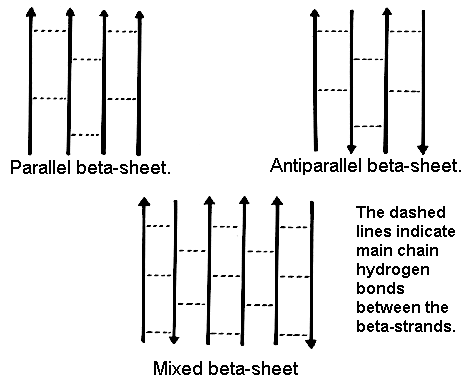
In parallel beta-sheets the strands all run in one direction, whereas in antiparallel sheets they all run in opposite directions. In mixed sheets some strands are parallel and others are antiparallel.
Below is a diagram of a three-stranded antiparallel beta-sheet. It emphasises the highly regular pattern of hydrogen bonds between the main chain NH and CO groups of the constituent strands.
In the classical Pauling-Corey models the parallel beta-sheet has somewhat more distorted and consequently weaker hydrogen bonds between the strands.
Beta-sheets are very common in globular proteins and most contain less than six strands. The width of a six-stranded beta-sheet is approximately 25 Angstroms. No preference for parallel or antiparallel beta-sheets is observed, but parallel sheets with less than four strands are rare, perhaps reflecting their lower stability. Sheets tend to be either all parallel or all antiparallel, but mixed sheets do occur.
The Pauling-Corey model of the beta-sheet is planar. However, most beta-sheets found in globular protein X-ray structures are twisted. This twist is left-handed as shown below. The overall twisting of the sheet results from a relative rotation of each residue in the strands by 30 degrees per amino acid in a right-handed sense.
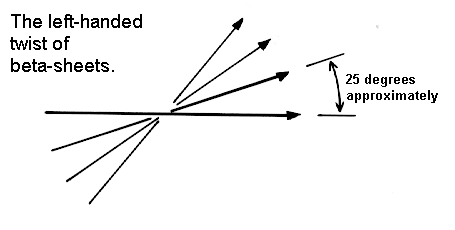
Parallel sheets are less twisted than antiparallel and are always buried. In contrast, antiparallel sheets can withstand greater distortions (twisting and beta-bulges) and greater exposure to solvent. This implies that antiparallel sheets are more stable than parallel ones which is consistent both with the hydrogen bond geometry and the fact that small parallel sheets rarely occur (see above).