 Click here for a
space-filling model of potato carboxypeptidase A inhibitor (38 residues long)
binding to the active site of bovine carboxypeptidase A. Also examine the
pdb file with RasMol.
Click here for a
space-filling model of potato carboxypeptidase A inhibitor (38 residues long)
binding to the active site of bovine carboxypeptidase A. Also examine the
pdb file with RasMol.
Back to Tertiary Structure Index
Consider a chemical reaction in which substrate S is converted to product P, by an enzyme E. Enzymes associate with their substrates to form an enzyme-substrate complex (ES). In the simplest model (the Michaelis- Menten model) E, the enzyme and S associate with a rate constant k1, while ES dissociates back to E and S with a constant k2, or forms E and P with a constant k3 (the turnover number). The least stable species on the reaction pathway is called the transition state. Catalysis involves lowering the energy of this transition state; as a result the rate of the reaction may be increased over a million-fold compared to the uncatalysed process. Enzymes bind their substrates more tightly in the transition state than in their normal states, ie the transition state ES is stabilized. For a detailed treatment of enzyme kinetics, refer to Fersht (1984).
A more detailed classification can be seen in the Swiss-Prot Enzyme Data Bank and the Enzyme Structures Database at UCL .
Only a small proportion of the enzyme's volume constitutes the active site, and therefore only a small fraction of the residues of the polypeptide chain are in contact with the substrate. The residues which constitute the active site are often not close to each other in the primary sequence, but the tertiary fold brings them close together in three-dimensional space. Active sites often involve residues on connecting loops between helices and sheets, rather than those which are part of these regular secondary structures. In many instances, active sites occur at the junction between two domains making tertiary contacts.
An enzyme molecule will be inactivated if the substrate can no longer bind to the active site. This may be effected by an inhibitor covalently bonding to the site, or binding very tightly so that its dissociation is very slow. This is irreversible inhibition (see the example of chymotrypsin below). In reversible inhibition, there is a rapid equilibrium of the enzyme and inhibitor. Competitive inhibitors bind to the active site. Non-competitive inhibitors bind elsewhere, but reduce the rate constant (the "turnover number") of the formation of enzyme E and product P from the ES complex. Effector molecules may act by having an opposite effect. In oligomeric enzymes with several active sites, allosteric inhibitors and effectors associating with one site affect the binding capabilities of others (see above on allosteric interactions).
Click here for a
space-filling model of potato carboxypeptidase A inhibitor (38 residues long)
binding to the active site of bovine carboxypeptidase A. Also examine the
pdb file with RasMol.
An example of this is citrate synthase (or "synthetase"), composed of alpha-helices as shown below.
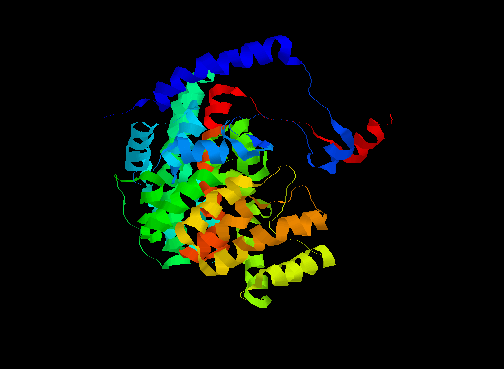
In one form, the surrounding domains are closed around the active site with both the product (citrate) and coenzyme (coenzyme A, or CoA) bound. In the open form, only citrate is bound (see below).
 Click here for the PDB structure with RasMol of the closed form (holoenzyme),
showing both citrate and CoA. The diagram on the left is obtained by
selecting Display:sticks and Colour:chain.
Click here for the PDB structure with RasMol of the closed form (holoenzyme),
showing both citrate and CoA. The diagram on the left is obtained by
selecting Display:sticks and Colour:chain.
 Click here for the PDB structure with of the open form (apoenzyme), with
citrate only.
Click here for the PDB structure with of the open form (apoenzyme), with
citrate only.
The function of the enzyme is to catalyze 1 of the reactions of the citric acid cycle, in which an acetyl group, covalently bound to CoA (in the form of acetyl-CoA) reacts with the substrate and 1 water moleculeto give citrate and CoA. CoA is a universal carrier of acyl (in many cases acetyl) groups. Acetyl-CoA has a high acetyl group transfer potential.
Click here for a diagram of the closed form of citrate synthase, indicating the structure of CoA (it consists of an ADP 3'-phosphate, a pantothenate and a 2-aminoethanethiol group, covalently bonded), and also including the citrate ion.
The coenzyme NAD+ (nicotinamide adenine dinucleotide) is involved in the function of certain dehydrogenases. The crystal structures of various enzymes of this type- (1) dogfish lactate dehydrogenase, (2) soluble porcine malate dehydrogenase,(3) horse liver alcohol dehydrogenase, (4) lobster glyceraldehyde-3-phosphate dehydrogenase, (5) Bacillus stearothermophilus glyceraldehyde-3-phosphate dehydrogenase (apo form) reveal that each consist of 2 domains, one of which, the NAD+ binding domain, is similar in each case. The other domain differs markedly; although (1) and (2) are very similar and appear to have evolved from a common precursor. Note that in some of these crystal structures, there is more than one enzyme molecule in the asymmetric group. In each case except the apoenzyme (5), selecting the Colour:chain option on RasMol highlights the bound nucleotide.
Manuel Peitsch provides two different views of the Bacillus stearothermophilus apoenzyme (5), and also several images of horse liver alcohol dehydrogenase:
The NAD+ binding domain is also present in other nucleotide- binding proteins, but also occurs in some proteins, eg flavodoxin, which do not bind nucleotides. Possibly this is simply a result of the finite number of ways in which a polypeptide chain may be folded, rather than being the product of divergent evolution.

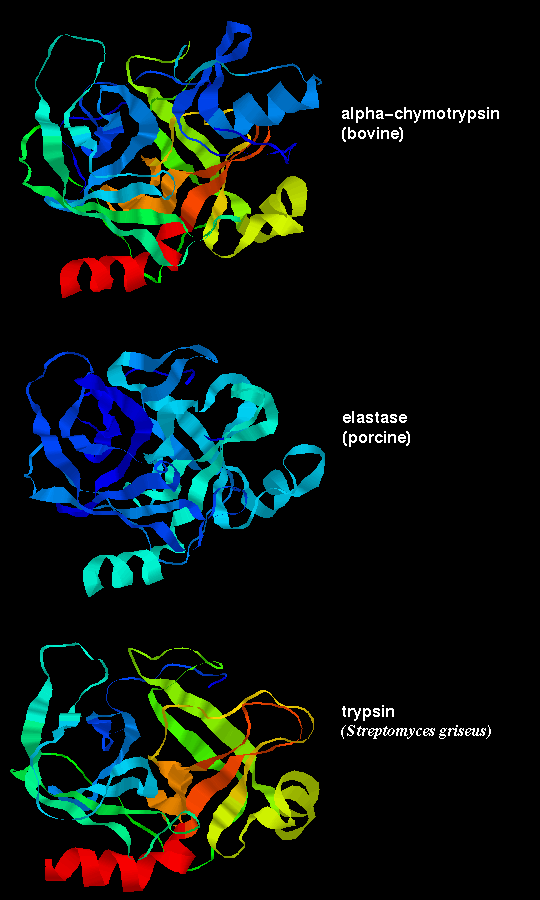
However, the sequences of the 3 proteins are only 50% homologous. Residues on the surface of the enzyme are only about 10% homologous, whereas the figure for buried residues, ie including the functionally important ones which contribute to the active site, is approximately 60%.
These three proteases are the result of divergent evolution. They differ in their specificity: chymotrypsin has a large pocket which accommodates the large hydrophobic side chains of Phenylalanine, Tyrosine and Tryptophan, and so catalyses the cleavage of peptides and esters of these amino acids. Trypsin has an Aspartate residue (189) at the bottom of the pocket (instead of Ser-189 as in chymotrypsin), and this Asp forms a salt bridge with the positively charged group at the end of the substrate Lysine and Arginine side chains, on which this enzyme acts. Elastase only accommodates small hydrophobic side chains eg Alanine, as the mouth of the pocket is partially blocked by the side chains of Val-216 and Thr-226 (these residues are both Gly in chymotrypsin). Examine Andrew Wallace and Roman Laskowski's LIGPLOT diagram of a tryptophan residue in the active site of chymotrypsin, and Manuel Peitsch's diagram of the enzyme highlighting the active site. There is a similar picture of elastase.
The serine proteases were so named as they have a highly reactive serine residue, Ser-195, which attacks the carboxyl group of the substrate.This results in an acylenzyme intermediate consisting of the substrate covalently bound to the enzyme at this serine.
However, the reactivity is dependent upon the arrangement of the serine side chain with two other polar side chains, approximately in a straight line, which is characteristic of all serine proteases. The Ser-195 is positioned at one end of this line, while at the other end is Asp-102, with His-57 in the middle. This is called the catalytic triad. Notice how far apart these three residues are in the sequence- the tertiary structure of the polypeptide chain brings them together in the required arrangement.
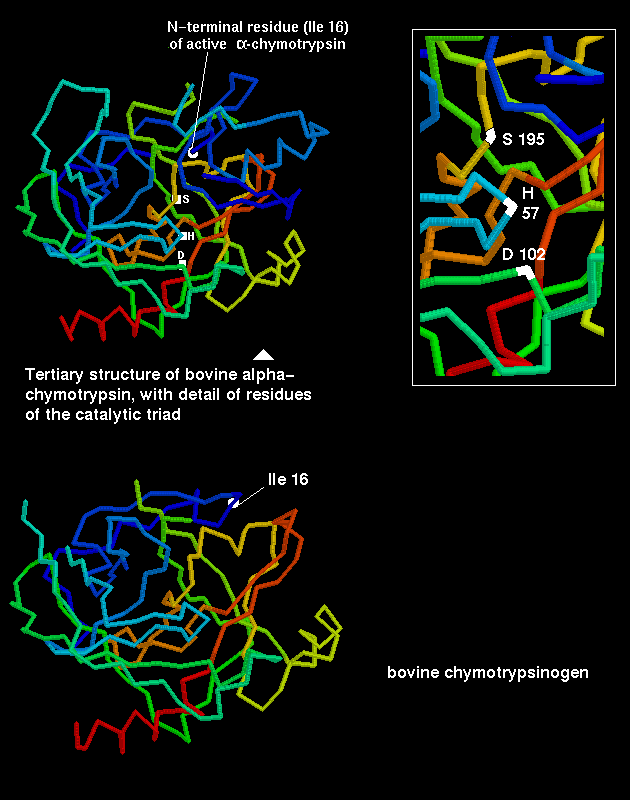 The catalytic triad is indicated in this diagram.
The catalytic triad is indicated in this diagram.
The residues of the catalytic triad form a charge relay network. His-57, polarized by Asp-102, acts as a proton shuttle which accepts the hydrogen ion from Ser-195 as it makes a nucleophilic attack on the substrate.
The inactive precursor (the zymogen) of chymotrypsin is the 245-residue protein chymotrypsinogen . A cleavage is made (by trypsin) between residues 15 and 16, to form an active form of the enzyme called pi-chymotrypsin. Further cleavages are made (by another pi-chymotrypsin molecule) to remove residues 14, 15, 147 and 148, to give the stable form of the enzyme, alpha-chymotrypsin. Note that trypsin also undergoes similar activation by means of cleavage. This is therefore a positive feedback mechanism, which activates the pancreatic enzymes in the intestine (the zymogens are secreted by the pancreatic cells).
The activation of the zymogen by cleavage involves highly localized conformational changes. Cleavage between the 15th and 16th residues forms an amino-terminal group on Ile-16, which turns inwards and interacts with Asp-194 in the interior of the molecule; this stabilizes the protein. This electrostatic interaction triggers other alterations in conformation, which result in the correct arrangement of the residues forming the cavity for the substrate; this cavity is not fully formed in chymotrypsinogen.
Differences between the structure of alpha-chymotrypsin and chymotrypsinogen
are indicated in the previous diagram.
Also examine Manuel Peitsch's diagram
highlighting the different position of Ile-16 in the enzyme and zymogen.
Some non-mammalian serine proteases have been found to have a very similar
tertiary structure to their mammalian counterparts, and are 20-50% homologous
with them; for example see
trypsin from Streptomyces in a
previous diagram. However, other non-mammalian
examples have no homology to the mammalian enzymes, and have completely
different tertiary structure; yet the same catalytic triad, and arrangement
of hydrogen bonding groups to the substrate, has evolved
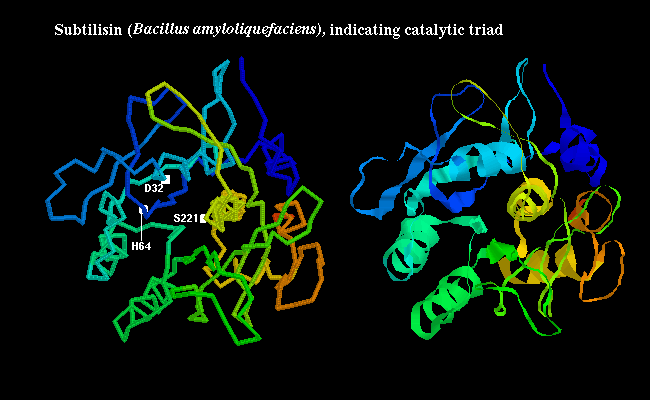
independently (convergent evolution). In the bacterial serine protease
subtilisin,
the triad consists of Ser-221, His-64 and Asp-32 as shown
below:
The molecular basis of the function of chymotrypsin will be examined in more detail in a future chapter.
Back to Main PPS Index
J. Walshaw
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}