Secondary structure elements are observed to combine in specific geometric arrrangements known as motifs or supersecondary structures. In this section we will look at motifs consisting of no more than three secondary structure elements. Larger motifs such as the greek key will be examined in the sections on tertiary structure and protein folds.
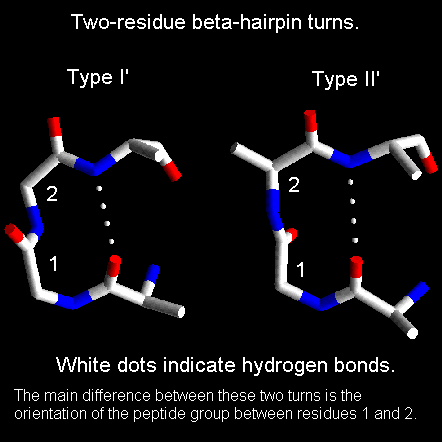
Beta-hairpins are one of the simplest supersecondary structures and are widespread in globular proteins. They occur as the short loop regions between antiparallel hydrogen bonded beta-strands. In general a reverse turn (or beta-turn, as it they are sometimes called) is any region of a protein where there is a hydrogen bond involving the carbonyl of residue i and the NH group of residue i+3. An alternative definition states that the alpha-carbons of residues i and i+3 must be within 7.0 Angstroms. The structures of reverse turns are outlined in the section on peptide geometry. In this section we will concentrate on those turns which occur between consecutive beta-strands, known as beta-hairpins. Sibanda and Thornton have devised a system for classifying beta-hairpins which is based on two conventions for defining loop regions. In this section we will not go into such details as the objective is indicate the most commonly observed hairpin loop structures.
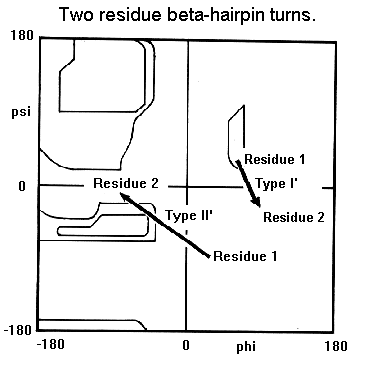
Beta-hairpin loops adopt specific conformations which depend on their lengths and sequences. Sibanda and Thornton have shown that 70% of beta-hairpins are less than 7 residues in length with the two-residue turns forming the most noticeable component. These two-residue beta-hairpins all adopt one of the classical reverse turn conformations with an obvious preference for types I' and II'. Type I 2-residue hairpins also occur but with lower abundance. This contrasts with reverse turns where types I and II tend to dominate. In beta-hairpins the type I' turn has the correct twist to match the twist of the beta-sheet and modelling studies indicated that if either type I or type II turns were to connect the antiparallel beta-strands, they would diverge within a short distance from the turn.
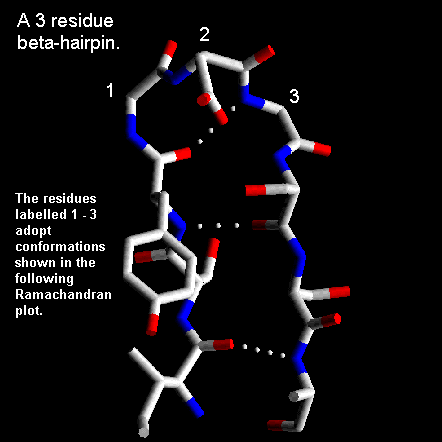
Normally the residues at the ends of the two beta-strands only make one hydrogen bond as shown below. The intervening three residues have distinct conformational preferences as shown in the Ramachandran plot. The first residue adopts the right-handed alpha-helical conformation and the second amino acid lies in the bridging region between between alpha-helix and beta-sheet. Glycine, asparagine or aspartate are frequently found at the last residue position as this adopts phi and psi angles close to the left-handed helical conformation. For RasMol-MIME, here's a pdb-file
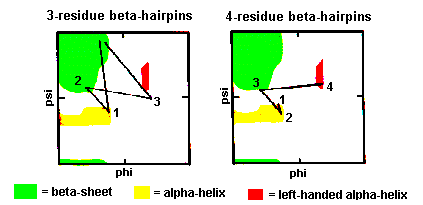
These are also quite common with the first two residues adopting the alpha-helical conformation. The third residue has phi and psi angles which lie in the bridging region between between alpha-helix and beta-sheet and the final residue adopts the left-handed alpha-helical conformation and is therefore usually Gly, Asp or Asn. Here's a coordinate structure file to view in RasMol.
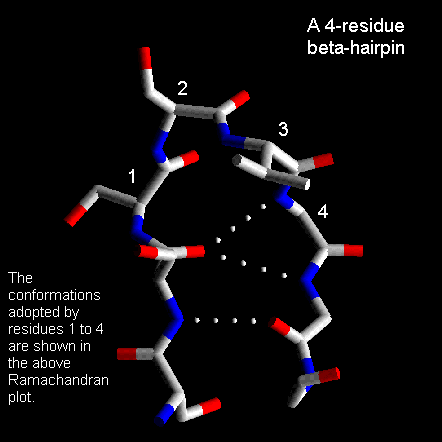
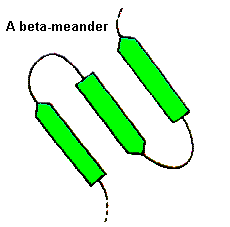
For these, a wide range of conformations is observed and the general term 'random coil' is sometimes used. Consecutive antiparallel beta-strands when linked by hairpins form a supersecondary structure known as the beta-meander.
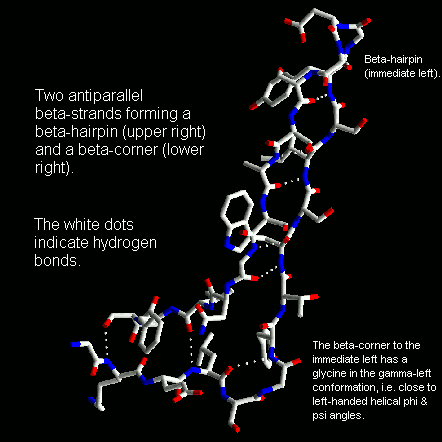
Beta-strands have a slight right-handed twist such that when they pack side-by-side to form a beta-sheet, the sheet has an overall left-handed curvature. Antiparallel beta-strands forming a beta-hairpin can accommodate a 90 degree change in direction known as a beta-corner. The strand on the inside of the bend often has a glycine at this position while the other strand can have a beta-bulge. The latter involves a single residue in the right-handed alpha-helical conformation which breaks the hydrogen bonding pattern of the beta-sheet. This residue can also be in the left-handed helical or bridging regions of the Ramachandran plot. Take a look at this example structure file from penicillopepsin (3APP) to examine with RasMol.
Beta-corners are observed to have a right-handed twist when viewed from the concave side.
A helix hairpin or alphaalpha-hairpin refers to the loop connecting two antiparallel alpha-helical segments. Clearly, the longer the length of the loop the greater the number of possible conformations. However, for short connections there are a limited number of conformations and for the shortest loops of two or three residues, there is only one allowed conformation. Antiparallel alpha-helices will interact generally by hydrophobic interactions between side chains at the interface. Therefore, hydrophobic amino acids have to be appropriately positioned in the amino acid sequence (one per turn of each helix) to generate a hydrophobic core. Efimov has analysed the conformations of alpha-alpha-hairpins and some of his results are summarised below.
The shortest alpha-helical connections involve two residues which are oriented approximately perpendicular to the axes of the helices. Analysis of known structures reveals that the first of these two residues adopts phi and psi angles in the bridging or alpha-helical regions of the Ramachandran plot. The second residue is always glycine and is in a region of the Ramachandran plot with positive phi which is not available to other amino acids.

Three residue loops are also observed to have conformational preferences. The first residue occupies the bridging region of the Ramachandran plot, the second adopts the left-handed helical conformation and the last residue is in a beta-strand conformation.
Four-residue loops adopt one of two possible conformations. One is similar to the three residue loop conformation described above except that there is an additional residue in the beta-strand conformation at the fourth position. The other conformation involves the four residues adopting bridging, beta, bridging, and beta conformations, respectively.
Short loop regions connecting helices which are roughly perpendicular to one another are refered to as alpha-alpha-corners. Efimov has shown that the shortest alpha-alpha-corner has its first residue in the left-handed alpha-helical conformation and the next two residues in beta-strand conformations. This conformation can only be adopted when the two helices form a right-handed corner. Indeed, if the helices were linked to form a left-handed corner there would be steric hindrance. This may explain the scarcity of left-handed alpha-alpha corners in protein X-ray structures.
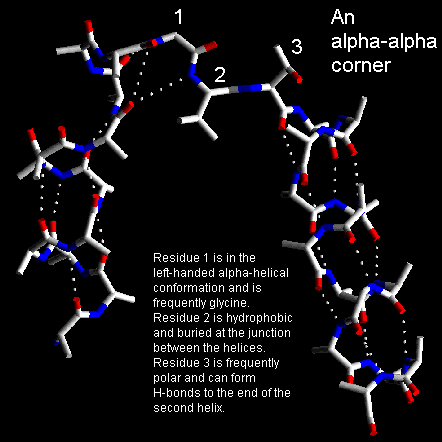
The C-terminal residue of the first helix, which is in the left-handed alpha-helical conformation, must have a short side chain to avoid steric hindrance and is observed commonly to be glycine. The first residue of the second helix, which is in the beta-conformation, frequently has a small polar side chain such as Ser or Asp which can form hydrogen bonds with the free NH groups at the amino-terminal end of the second helix. The central residue of the alpha-alpha-corner is almost always hydrophobic as it is buried and interacts with other non-polar side chains buried where the ends of the two helices contact each other.
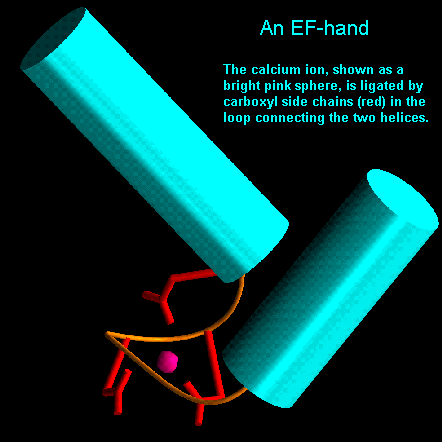
The loop regions connecting alpha-helical segments can have important functions. For example, in parvalbumin there is helix-turn-helix motif which appears three times in the structure. Two of these motifs are involved in binding calcium by virtue of carboxyl side chains and main chain carbonyl groups. This motif has been called the EF hand as one is located between the E and F helices of parvalbumin. It now appears to be a ubiquitous calcium binding motif present in several other calcium-sensing proteins such as calmodulin and troponin C.
EF hands are made up from a loop of around 12 residues which has polar and hydrophobic amino acids at conserved positions. These are crucial for ligating the metal ion and forming a stable hydrophobic core. Glycine is invariant at the sixth position in the loop for structural reasons. The calcium ion is octahedrally coordinated by carboxyl side chains, main chain groups and bound solvent..
A different helix-loop-helix motif is also common to certain DNA binding proteins. This motif was first observed in prokaryotic DNA binding proteins such as the cro repressor from phage lambda. This protein is a homodimer with each subunit being 66 amino acids in length. Each subunit consists of an all-antiparallel three stranded beta-sheet with three helical segments inserted sequentially between the first and second beta-strands. The two subunits of cro associate by virtue of the third beta-strands which interact forming a six-stranded beta-sheet in the centre of the molecule. Mutagenesis and biochemical work had indicated that residues in the second helix of each cro monomer interacted with DNA. Accordingly model building studies indicated that both these helices in the dimeric protein would fit into the major groove of B-DNA. These proteins recognise base sequences which are palindromic, i.e. possess an internal two-fold symmetry axis. The two recognition helices of the cro protein are also related by a two-fold axis passing through the central beta-sheet region of the dimer. Therefore, the recognition helices of the cro dimer fit into the major groove of the DNA and interact with each identical half of the palindrome. Hence, the second helix of the helix-turn-helix motif has an important role in recognising the DNA while the remainder of the structure serves to keep the two helices in the correct relative position for fitting in the major groove of DNA. Many other helix-turn-helix proteins with different folds exhibit essentially the same mode of binding to DNA.
Antiparallel beta-strands can be linked by short lengths of polypeptide forming beta-hairpin structures. In contrast, parallel beta-strands are connected by longer regions of chain which cross the beta-sheet and frequently contain alpha-helical segments. This motif is called the beta-alpha-beta motif and is found in most proteins that have a parallel beta-sheet. The loop regions linking the strands to the helical segments can vary greatly in length. The helix axis is roughly parallel with the beta-strands and all three elements of secondary structure interact forming a hydrophobic core. In certain proteins the loop linking the carboxy terminal end of the first beta-strand to the amino terminal end of the helix is involved in binding of ligands or substrates. The beta-alpha-beta motif almost always has a right-handed fold as demonstrated in the figure.
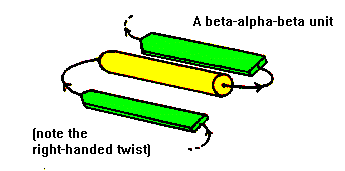
The subsequent sections of this course on protein folds and domains will demonstrate how the beta-alpha-beta motif is an important and widespread element of supersecondary structure.
Introduction to Protein Structure, C.Branden and J.Tooze, Garland, New York (1991).
A novel super-secondary structure of proteins and the relation between the structure and amino acid sequence, A.F.Efimov, FEBS Lett. 166, 33-38 (1984).
Structure of alpha-alpha-hairpins with short connections, A.V.Efimov, Protein Engin.4, 245-250 (1991).
Structure of beta-beta-hairpins and beta-beta-corners, A.V.Efimov, FEBS Lett., 284, 288-292 (1991).
Conformation of beta-hairpins in protein structures, B.L.Sibanda, T.L.Blundell and J.M.Thornton, J.Molec.Biol. 206, 759-777 (1989).
Beta-hairpin families in globular proteins, B.L.Sibanda and J.M.Thornton, Nature 316, 170-174 (1985).