Contributed by Antti Iivanainen, University of Oulu, FINLAND
The coiled-coil structure is used by nature to stabilize alpha helices in proteins. This is possible through a very efficient burial of hydrophobic side chains so that for the most part, they are not accessed by polar water molecules. As a result of this structure the helices are quite stable. In fact, many structural proteins both inside and outside of cells (keratins, tropomyosin, laminin etc.) that have to bear considerable stress have a coilel-coil domain. There seems to be a couple of basic essential features that are common to coiled-coil peptides. First, the overall secondary structure is alpha helical and secondly, the hydrophobic residues are arranged on one side of the helices. The typical positioning of the hydrophobic residues in coiled-coils, the coiled-coil motif, can be often recognized from primary structure of the protein.
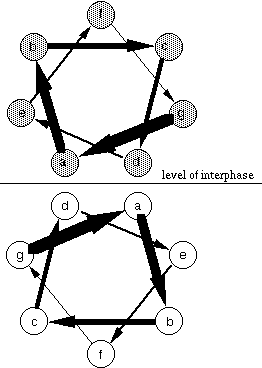
A common way of illustrating the amphipathic nature of helices is to use a helical wheel represention, in which the helices are viewed from the end.
Helical wheel
From this view, the logic in the identification of coiled coil motifs from the primary sequence alone becomes apparent. It takes about 3,5 residues to make a complete turn around an alpha helix. Therefore, from the helical wheel projection, there are 7 different positions for amino acid residues along the circumpherence of the helix. Looking from N-terminus, these are labelled from a to g in a clockwise manner so that the residues in positions a and d form the hydrophobic interphase between helices. However, there are actually slightly more than 3.5 residues per one turn. This creates a slight (0-15 degrees when compared with helix axis) inclination of the hydrophobic stripe by the residues in a and d positions. Therefore, the helices cross at an angle between 0-30 degrees. From this, it follows that the right-handed helices cannot form an apolar interphase of several turns' lenght unless the superhelix is turned anticlockwise (making it left-handed). The helices can be parallel or antiparallel. The former is common when the helices belong to different polypeptides and the latter prevails in coiled coils made up from a single polypeptide chain. Also, the number of helices in the coil vary usually between two and four.
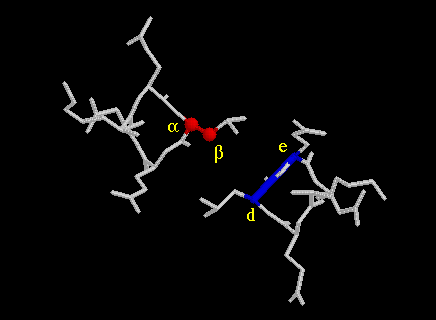
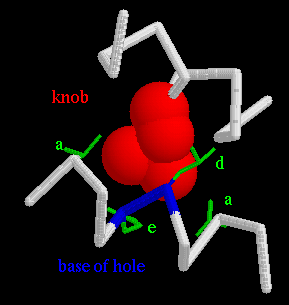
The hydrophobic side chains in positions a and d buried into the neighbouring helix in a way that was proposed in 1953 by Crick (3). He called it knobs-into-holes packing. In this model, the side chains in positions a and d of one helix are considered as knobs, which pack into the holes lined by the residues in the neigbouring helix. This is elegantly and simply illustrated by helical net diagram. It shows a projection of the two interacting helices on interphase level. An illustration from GCN4 coiled coil (5) (not a helical net diagram!) of a Leu19 knob fitting snugly into a hole can be seen here. (a, d and e denote position of residues in the coiled coil motif.) For a parallel dimer, a knob in position a (at residue n) fits into the hole in the other helix that is lined in clockwise sequence (when looking into the hole) by residues in positions a(n), d(n), g(n-7) and d(n-7). Accordingly a knob at d(n) is in the hole lined by d(n), a(n), e(n) and a(n+7).
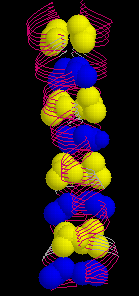
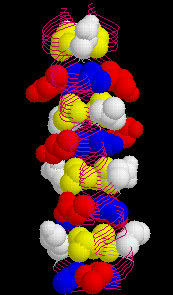
The wild type peptide was shown to adopt a parallel, dimeric coiled coil which formed a twisted elliptical cylinder (about 45 Å x 30 Å ). In this cylinder, a hydrophobic core can be distinguished. It is made up from alternating layers of residues from positions a or d. (Here is an image of the wild type GCN4 leucine zipper with the d-layer colored yellow and a-layer blue, respectively.) Also, residues from positions e (white) and g (red) which often contain hydrophobic methylene groups in the side chain take part in the formation of the hydrophobic core.
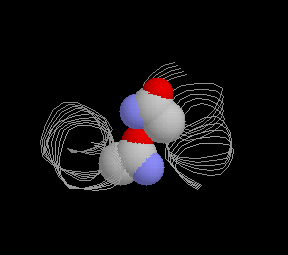
The specificity of the interhelical contact is largely determined by residues in the positions e and g as shown by elegant construction of chimeric peptides (9). In several coiled coil proteins, these positions are occupied by residues with polar and ionic side chains enabling, in many cases, specific ionic interactions between the contacting helices. Also, in some cases, there are polar residues in positions a or d which - if matched from the counter helix - create a specific and relatively strong hydrophilic component within the hydrophobic interphase. For example, in the GCN4-p1 asparagine-16 forms a hydrogen bond in the core with Asn16 from the other helix (5) (gif.image here ). In an analogous position, the c-fos/c-jun dimer contains lysine (K176 in c-fos) and asparagine (N291 in c-jun) at a-positions, which are interacting with the g-residues of the other helix, that is glutamine 290 at g-position of c-jun and glutamic acid 175 at g-position of c-fos (See fig. 3d in a fabulous work by Glover and Harrison, ref#10).
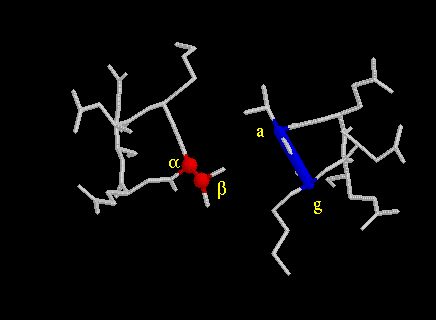
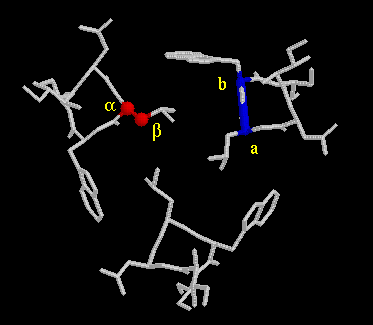
By analyzing a number of different mutated GCN4 peptides, Kim, Alber and their coworkers were able to distinguish different packing geometries (7,8). Below are images that demostrate the principles of the packing geometry. Perpendicular and parallel geometries are from GCN4-p1 at Leu5 (d-level) and Val9 (a-level), respectively. The acute geometry is from influenza haemagglutinin at Leu91 (a-level) (11-12).
perpendicular
parallel
acute
In summary, these packing geometries are defined by the angle which the C(alfa)-C(beta) bond of the knob (red) forms with the C(alfa)-C(alfa) vector (blue) at the base of the hole on a projection from the end of the coiled helices.
From exhaustive analysis of the different peptides (7,8), it could be concluded that in the GCN4 coiled coil model peptide, the packing geometries are largely determined by the number of interacting helices and the overall tertiary structure. For example, the a-layer packs into a parallel geometry (see above) in the dimer but into a perpendicular geometry in the tetramer. In the trimer, both layers pack into acute but different geometries. On the other hand, ß -branched residues (Val and Ile) are disfavored in the perpendicular geometry possibly because they would have to adapt a rare rotamer. Also, isoleucine is favored over leucine in parallel geometry (0,4 kcal/mol) thus increasing the likelyhood of dimerization over tetramerization with ß -branched isoleucine in the a-layer but not in the d-layer. Therefore, in the GCN4 case, the placement of beta-branced residues into a- and d-positions in the primary structure determined to a large extent the number of helices forming the coiled coil. Furthermore, in light of the analysis of the regulation of c-fos/c-jun dimerization by using chimeric peptides, the placement of specific residues at e- and g-positions mostly determine which proteins dimerize into a functional transactivator. Perhaps this mechanism could be applied also to other bZIP DNA-binding proteins. Hence, for example, these residues could have a major role the recognition of a specific combination two half sites in a given stretch of DNA. (Click here for an image of GCN4 complexed with its target, AP1 (13).)
Influenza virus haemagglutinin (HA) is a viral membrane glycoprotein essential for viral infectiviry. HA is synthesized as a precursor (HA0) which is then cleaved into two, disulfide linked peptides HA1 and HA2. Native HA is a trimer comprising three HA1-HA2 subunits. Trimerization is mediated through a coiled coil. Native HA is not capable of promoting fusion of viral and host cell membranes but the fusogenic HA is irreversibly activated by either a mild acid treatment in the endosomes (pH=5) or by heatshock to above 60 degrees C. Native HA is cleaved by bromelain and the resulting ectodomain (BHA) is functional in terms of acid activated membrane fusion activity.
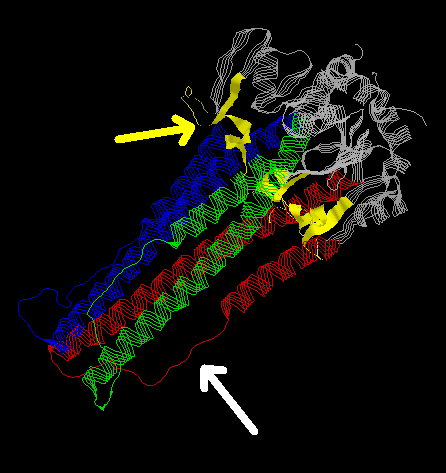
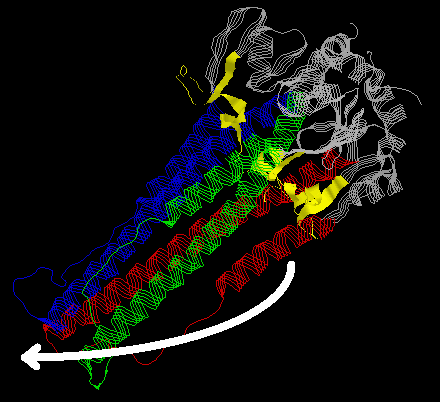
The three-dimensional structure of HA2-trimer in the native state (11,12) shows how the N-terminal fusion peptide (yellow) is inaccesible. (The viral membrane is on the top right hand corner of the image.) Also, there are two helices in each monomer (blue, red and green) that are separated by a loop structure (white arrow). The native form of HA2 is stabilized partially through the interaction between the long and the short helices but nevertheless, this form is not completely stable. A major rearrangement of structure takes place during fusogenic activation (14,15).
In a very elegant study, Chavela M. Carr and Peter S. Kim characterized the structural dynamics of the loop peptide using CD spectroscopical and sedimentation equilibrium analysis (16). They showed that at neutral pH the peptide is monomeric and in a random conformation. However, at pH4.8 it is a trimer with >90% alpha helicity. The primary sequence corresponding to the loop structure is consistent with an extended coiled coil motif continuous with the motif on both flanking helices. At neutral pH, however, folding into an alpha-helix is likely to be hindered by strong electrostatic repulsive forces from charged glutamic acid residues at consequtive e-positions within the coiled coil motif. On the other hand, if the folding of HA0 is guided to a local energy minimum (with the short and long helices packed against each other), then raising the temperature to over 60 degrees C could provide enough energy to to procede with folding until a true energy minimum is reached. Thus, the hypothetical structure at the fusogenic state would consist of a continuous, long trimeric coiled coil containing also those residues that fold into the loop structure at neutral pH. This would trust forward and expose the fusogenic peptide.
Back to the main index
Back to the course backbone.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
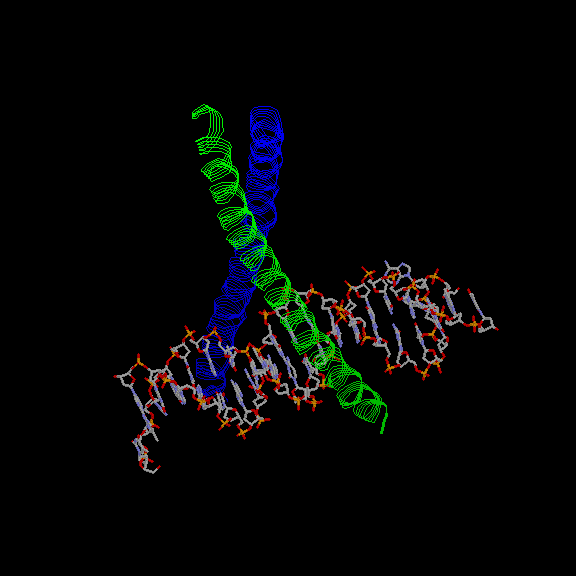{kind=link}
{kind=link}
{kind=link}