Immunoglobulins are naturally designed glycoproteins that are extremely versatile in binding with high affinity and specificity with an estimated one million antigenic epitopes. It is not surprising that the Ig molecule demonstrates many salient features of protein structure and function. Each divalent Ig consists of two pairs of light and heavy chains. Each chain is composed of discrete domains connected by flexible linkages. The light chain consists of one variable domain VL and and one constant domain CL. The heavy chain has one variable domain VH and and 3 constant domains CH1, CH2 and CH3.
The elbow junction of Ig connects the variable domain to the constant domain. The VL and CL domains are linked by an extended strand, as are the VL and CH1 domains. VL and VH pack together, CL and CH1 doing similarly, each pair forming a immunoglobulin fold of 2 antiparallel beta-sheets. The VL-VH dimer freely rotates relative to the CL-CH1 dimer over a range of about 500, like an elbow joint.
The ball of the V-C elbow joint is made of two residues in the CH domain, whilst the socket is made of three residues in the VH domain. The joint motion is mainly a hinge motion, enriched with a shear motion less constrained by close packing of interdigitating side chains. The socket residues can move up to 4.5 Angstroms relative to the ball residues.
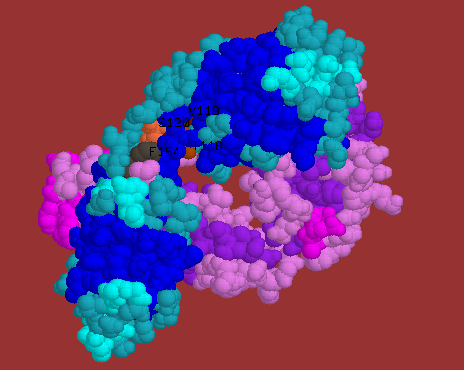
![]() Human
Fab 8FAB(Fab from an IgG1 monomer) elbow joint with the ball and socket
in van der Waals balls. Socket residues H118, H119 and H120 in CPK colouring.
Ball residues are H154(dark grey) and H155(light grey). The rest of the
molecule is shown in ribbon model. Heavy chain sheet in blue. Heavy chain
helix in cyan. Light chain sheet in purple. Light chain helix in magenta.
Turns and extended strands or loops are violet in the light chain and greenblue
in the heavy chain.
Human
Fab 8FAB(Fab from an IgG1 monomer) elbow joint with the ball and socket
in van der Waals balls. Socket residues H118, H119 and H120 in CPK colouring.
Ball residues are H154(dark grey) and H155(light grey). The rest of the
molecule is shown in ribbon model. Heavy chain sheet in blue. Heavy chain
helix in cyan. Light chain sheet in purple. Light chain helix in magenta.
Turns and extended strands or loops are violet in the light chain and greenblue
in the heavy chain.
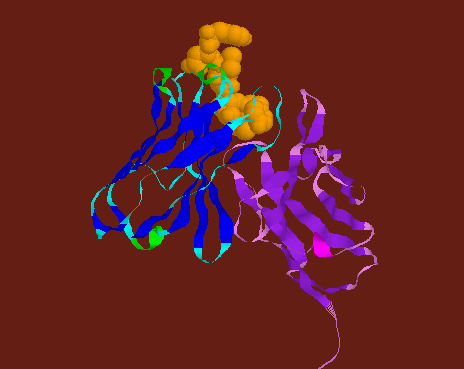
![]() Human
Fab (Fab from an IgG1 monomer) elbow joint in spacefill model.
Human
Fab (Fab from an IgG1 monomer) elbow joint in spacefill model.
Each Fab fragment contains one antigen-binding site formed by 3 light chain hypervariable loops and 3 heavy chain hypervariable loops held in position by the VL-VH fold. According to the traditional lock-and-key model, each antibody type has a unique hypervariable region that is specially shaped for its epitope. Upon engagement the key-and-lock is held in place by hydrogen bonds, salt bridges and van der Waals forces. Recently higher resolution crystallographic data of antibody-antigen complexes led to the suggestion that the interaction may be better viewed as an induced-fit model.
Rini et al examined the crystal structures of bound and unbound Fab 17/9 to a peptide immunogen from influenza virus haemagglutinin, HA1(75-110). Loop H3 of the antibody was noted to rearrange in order to accomodate TyrP105 of the bound peptide. The change involves a twisting of the two strands of the loop abount the long axis of the loop, creating a binding pocket for the beta turn of the peptide. The contact area between the peptide and antibody is large, about 400 square Angstrom.
![]() CDR
loops in Fab. H3 loop is shown in red.
CDR
loops in Fab. H3 loop is shown in red.
{kind=link}
{kind=link}
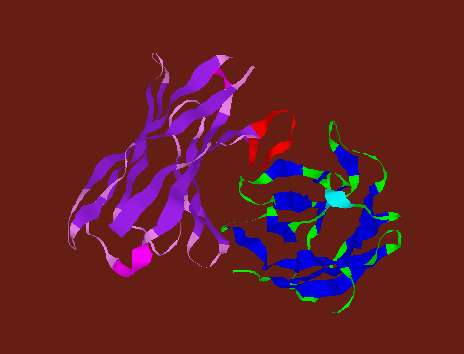{kind=link}
{kind=link}