Consider the range of interactions that must occur between DNA and proteins during the development from a human sperm and egg, which contain quiescently packaged DNA, to an adult with differential gene activity in a multitude of different tissues. Certain genes must be expressed at a precise time during development in a particular type of cell. Other genes must be expressed continually, or perhaps at one particular time during the cell cycle. The expression of the genetic information is controlled predominantly through the interaction of proteins with DNA. Some proteins must bind DNA in a sequence-dependent fashion, but some do it in a sequence-independent fashion. A variety of proteins are involved in the replication and repair of DNA (Table 1).
Table 1
| Proteins which interact with DNA |
|---|
| 1. REPLICATION AND REPAIR. |
| a. DNA polymerases of different types |
| b. DNA unwinding proteins and others in the replication complex |
| c. Ligases and repair proteins |
| d. Nucleases and excision enzymes of the repair process |
| 2. DNA PACKAGING PROTEINS. |
| a. Chromatin: nucleosomes and histones |
| b. Proteins of the sperm, protamines and other proteins |
| c. Virus condensation proteins: internal and coat |
| 3. TRANSCRIPTION. |
| a. RNA polymerase and its various subunits |
| b. Repressors and regulation of the initiation of transcription |
| c. Cyclic AMP receptor proteins (CRP) |
| d. Rho factors in the termination of transcription |
| 4. NUCLEASES. |
| a. Restriction endonucleases |
| b. Exo- and endonucleases |
| c. Nucleases of hydrolisis |
| 5. DNA MODIFYING PROTEINS. |
| a. Methylases |
Many of the proteins associated with DNA in the cromossomes of higher organisms may be presumed to influence both the structure and the function of the genetic material.
The conformation of DNA is influenced both by its sequence and by the counterions and other small molecules (in particular water) which interact with it. We will review the effect on DNA of basic proteins as counterions.
The most favorable interactions between amino acids and bases are for arginine and lysine with guanine, and also for lysine with thymine. These strongest preferences can be explained by simple considerations of partial charge interactions between amino acid side chains and base functional groups in the major groove (Suzuki,1994). It is interesting that elecrostatics interactions are of critical importance although the flexibility of the side chains may also be an additional contributing factor in the interactions.
Hydrogen bonding.
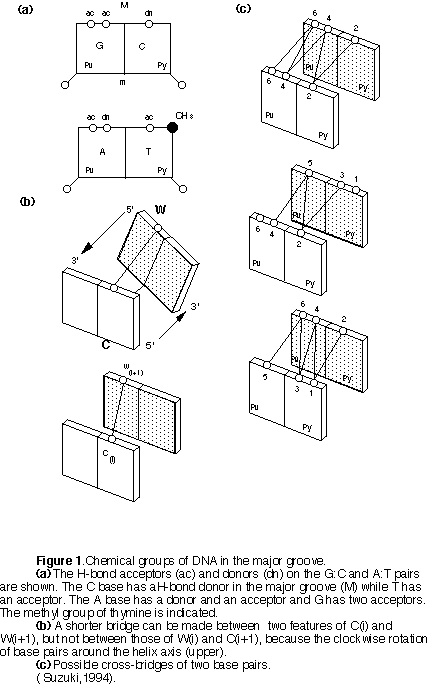
Contacts between amino acid residues and DNA bases are achieved by H-bonding or hydrophobic packing interactions. The H-bond donors and acceptors in the major groove are illustrated in figure 1.
These chemical groups are available for contact through H-bonds. Amino acid residues Glu and Asp have H-bond acceptors but not donors, therefore they can form H-bonds only with the C or A bases. Residues such as Ser, Cys, and Thr have a hydroxyl or sulphydryl group which can be used simultaneously as a donor and as acceptor, and they can therefore contact any base. Amino acid residues such Arg and Lys have H-bond donor but no acceptor group in their side-chains and therefore cannot contact the C base because it has no acceptor.
Hydrophobic interactions.
The main target for hydrophobic interaction in the major groove of DNA is the single methyl group of the T base. Hydrophobic residues such as Ala, Val, Ile, Leu, Met, Phe, Tyr, Trp and Thr can recognize thee methyl on T. The two ring CH groups of the C base are also possible targets for recognition by hydrophobic residues. However, interaction with the hydrogen atoms of C will not be as strong as with the methyl group of the thymine. Ala appears to be insufficiently hydrophobic to contact the C base.
The stem of the side chains of polar residues can also contact the methyl group of T.
"Bidentate" interactions.
Some residues can recognize two chemical groups on the same DNA base and such interactions would be stronger than single contacts. For example, Lys and Arg can make two H-bond contact to G, and Asn and Gln can contact A through two H-bonds, one
from A and the other to A. His has both a H-bond donor and a H-bond acceptor which point in almost opposite directions. Thus, only one of the two groups seems likely to be available for base recognition at any one time making a bidentate contact difficult.
Ionic effects on contacts.
Theoretically, Arg and Lys can bind to any of the bases, other than C, by H-bond. However, these two residues almost always bind to G even through a single contact. This is probably due to ionic effects on the contacts. The G base has two acceptors and a partial negative charge, while T base is less negative, as it has only one acceptor. The A base has an acceptor and a donor and is nearly neutral, therefore Arg and Lys bind to G much more often than to T, and to T more often than to A. His is less charged than Arg and Lys and thus its binding preference might be expected to be weaker.
Bridging of two bases by the same residue.
Sometimes two residues bridge two consecutive base pairs steps. The residues that bridge two base-pairs by H-bonds can be classified into three groups: the double acceptor type, the double donor type, and the acceptor donor type (Table 2).
Table 2.Possibility of complex formation by two hydrogen bonds
| Neutral Form | Ionized Form | ||||||
|---|---|---|---|---|---|---|---|
| Asp Glu | Asn Glu | Ser Thr Tyr | Cys | Asp Glu | Arg | ||
| A | + | + | + | + | |||
| G | + | + | + | + | + | + | |
| T | + | + | + | + | |||
| C | + | + | + | + | + | ||
(From Lancelot,G. 1977).
The polyelectrolyte character of a polymeric nucleic acid makes a large contribution to both the magnitude and the salt concentration dependence of its binding interactions with simple oligocationic ligands (Zhang,1996).
Short oligonucleotides are oligoelectrolytes, which differ strikingly in their interactions with salt ions from the behaviour of polyelectrolytes.The polyelectrolyte character of polmeric DNA has been proposed as the primary origin of the large dependences on salt concentration observed for the binding both of simple oligocations and of locally cationic surface regions of proteins to polymeric DNA.
Under typical experimental conditions (including physilogical salt concentrations) the effective range of electrostatic interactions is long enough so that many pariwise
interactions contribute to the electrostatic potential acting on the surface of DNA. Consequently, specification of the precise location of each charge is relatively less significant.
It has been seen that most of the ligand-DNA complexes were dissociated as the salt concentration increased. The binding to polymeric DNA is dominated by the release of cations that had been thermodynamically associated with DNA.
Basic peptides studied up to now have three different types of behaviour Campos et al,1986):
a)Stabilization of the B form. The B form has an intrinsic stability, which does not depend much on the precise structure of the counterion present. All of these several counterions contain several positive charges, as well as being suitable for hydrogen bonding. These counterions may substitute for water upon dehydration, so that the B form is maintained in the absence of water.
b)Alteration of the B form. The large peptides alter DNA conformation. This behaviour is probably due to the fact that large, charged peptides may have their conformation fixed upon interaction with DNA as a result of adapting themselves to the charge distribution of DNA, so that they are unable to play the same role as water molecules in stabilizing the B conformation of DNA.
c)Destabilizing of the B form. In a few cases the conformation of DNA is disrupted.The destabilization is probably due to the hydrophobic nature of the peptides, which interfere with the intramolecular interactions in the DNA molecule. These peptides probably alter the conformation of the phosphodiester chain through a complex interaction with DNA molecule.
Protamines may show a large compositional variety, although they always have a high concentration of basic amino acids.
More than 100 years ago protamine-DNA complexes were the first objects for scientific analysis of protein-nucleic acids complexes. These are specific to the male germ cell line. The main reason for replacement of histones by protamines during spermatogenesis appears to be the fact that protamines convert DNA molecules into a very compact state. It is expected that the main binding to DNA comes from electrostatic interactions (Porschke, 1991).
The structure of nucleoprotamine complexes has been studied by X-Ray diffraction.
The phosphate charges of the DNA present in the nuclei of eukaryotic organisms are always neutralized by proteins. This neutralization is almost complete in those sperm nuclei which contain protamines, so that the basic amino acid/phosphate ratio is close to
unity in such cells. This neutralization results in a complete insolubilization of the complex and only a relatively small change in its packing denstiy is observed when the humidity of the sample is modified. In fact nucleoprotamine sample only accepts about 25% of its dry weight as water of hydration at 100% relative humdity (Fita elt al,1983).
In contrast, the DNA in somatic nuclei is not fully neutralized by histones. Each core has a net positive charge about 20%, so that the eight molecules in a nucleosome core only neutralize the equivalent of 80 base pairs of the 146 present in a core particule. In fact it has been shown that core particles can accept a much larger amount of histones. The addition of histone H1 does not allow complete neutralization of the complex either. In fact external histones also bind to isolated chromosomes or nuclei. The relatively low degree of neutralization of DNA by histones makes the nucleohistone complex very sensible to the ionic conditions and to the presence of divalent ions.
It is possible that protamines besides of neutralizing all the phosphate charges with the basic residues also accomodate the neutral residues on the wide groove without steric hindrances.
It is generally accepted that the globular part of histone H1 interacts with the nucleosome core, whereas the highly charged C-terminal region neutralizes part of the phosphates found in internucleosomal DNA. So and so, Subirana et al. have been analysed the charge distritbution in this histone region .They have found that the percentage of positively charged residues varies significantly in different H1 histones, between 30 and 44%. In most cases the charge is uniformily distributed throughout the C-terminal region of histone H1.They concluded that this region may be considered as a featureless basic polypeptide with a rather costant charge density which reduces the charge of nucleohistone complex in a smooth way, thus allowing the formation of a higher order structure in chromatin fibers.
Histone H1 has a variable net charge but alway lower than 60+. Assuming that there is one molecule per nucleosome, histone H1 only neutralizes part of the charges of the DNA in the core and internucleosomal DNA, so that in the complete nucleohistone complex only about 60% of the phosphate charges in DNA are neutralized by histones. The other 40% are neutralized by inorganic and low molecular weight ions.
Of particular interest is the suggestion that the C-terminal region contains helical regions. In fact it was found that some synthetic polypeptides rich in alanine and lysine do interact with DNA in a helical conformation
It is suggested that the C-terminal region of histone H1 may interact with bundles of internucleosomal DNA and thus stabilize the higher order structure of nuleosomes in the
30 nm chromatin fiber, possibily in a zig-zag arrangement . The main difference with the protamine molecules will be that the latter have a much higher charge density (60 to 80%)
and allow a complete neutralization of DNA phosphates in the complex. Locally, the interaction may be quite similar in both cases, so that the helical regions of histone H1 may interact with DNA in a way similar to that shown for protamine.
As a general feature of all polypeptide-DNA complexes that have been studied, the B form of DNA is observed at high levels of relative humidity, with close to ten base-pairs per turn. Upon dehydratation of the complexes that have their phosphates charges fully neutralized by lysine side-chains, the A form is never observed. Only when the phosphate charges are neutralized in part by lysine side-chains and in part by sodium ions is the A form observed upon dehydratation. This behaviour has been found in complexes of DNA with protamines and with arginine oligopeptides (Azorin,1985) .
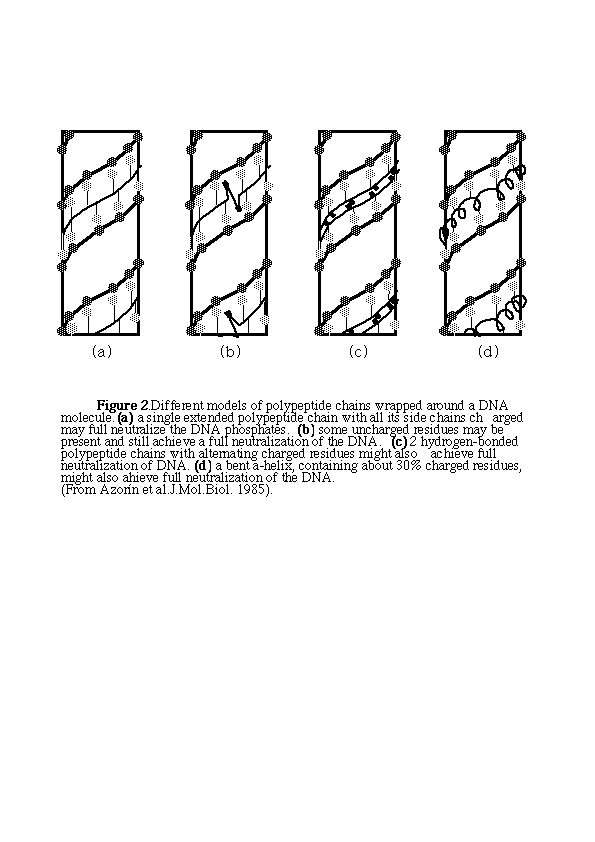
The DNA charges may be neutralized by polypeptide chains that run on one of the grooves of the DNA double helix. Some of the possible regular arrangements are indicated in figure 2.
So lysine-rich polypeptides with different conformations may interact with DNA, while their conformation may be either maintained or altered upon interaction. It is concluded that the same DNA sequence can be recognized by rather different sets of amino acid side-chains organized in different spatial arrangements. It appears that the lysine side-chains are rather mobile and allow an interaction with DNA without any main-chain conformational changes in either protein or DNA, as long as the charge density of the two interacting macromolecules has a similar value.
In the DNA-arginine complex the arginine molecules are located on the major grooves of DNA (Fita, 1983). The arginine molecule has a large hydrogen-bonding capability, and it may substitute for water if hydrogen bonds with other molecules are required to stabilize the B form of DNA. This residue may fill all the space available and in this way prevent a deformation of the DNA conformation. The arginine-rich polypeptides would wrapped around DNA in the same form as lysine-rich polypeptides. It was observed that the guanidinium group of arginine can form ion pairs with the phosphate groups although the interaction between carboxylate and arginine is stronger than between phosphate and arginine.
In many cases the degree of hydration of the DNA complexes has a strong influence on the conformation of DNA (Campos et al,1986). The mirror groove has a strong helical potential for the interaction and it is likely that some counterions tend to accumulate inside this groove.
However, it has been shown that arginine peptides interact with DNA in the major groove. It appears, therefore, that the precise site of interaction of a given peptide with DNA will depend on all the molecular interactions involved and will vary in each case.
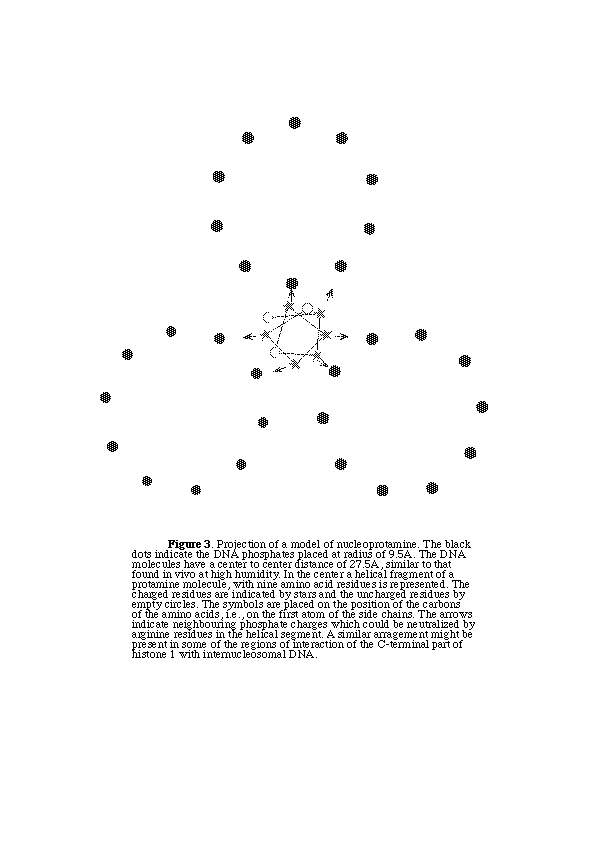
A possible model for the nucleoprotamine complex is based on the assumption that a significant lenght of the protamine molecule (40-60%) may adopt a helical form upon interaction with DNA. Each protamine molecule would be placed among three DNA molecules figure 3 . So that the axis of protamine is roughly parallel to the axis of the DNA molecules. If we consider a hypothetical protamine with 30 amino acids and 20 charged residues, it should run about 2 turns of the DNA helix (68A). Under these conditions a protamine molecule would neutralize 6.7 charges of each of the surrounding DNA molecules and vice versa, thus achieving complete charge neutralization. The protamine molecule would be about 50% helical and 50% partially extended, in order to span the required 68A. It has been observed that the higher the charge of the polypeptide, the lower the amount of a helix which would be allowed by the model, since the polypeptide chain should become fully extended in order to neutralize the phosphate charges (Subirana,1990).
-Azorín,F.(1985) Interaction of DNA with Lysine-rich polypeptides and proteins.J.Mol.Biol., 185, 371-387.
-Campos,L.(1986) Influence of Amino Acid and peptide counterions on the conformation of DNA.J.Mol.Biol., 187, 441-447.
-Fita, I.(1983) X-Ray diffraction study of DNA complexes with Arginine peptides and their relation to nucleoprotamine structure.J.Mol.Biol., 167, 157-177.
-Lancelot,G.(1977) Models of interaction between nucleic acids and proteins. Hydrogen bonding of Arginine with nucleic acids bases, phosphates groups and carboxylic acids.
-Lancelot,G.(1977) Selective recognition of nucleic acids by proteins:the specificity of guanine interaction with carboxylate ions.Proc.Nat.Acad.Sci.,74, No11, 4872-4875.
-Lustig,B.(1995) Consistencies of individual DNA base-amino acid interactions in structures and sequences. Nucleic Acids Research, 23, No 22, 4707-4711.
-Jones, C.(1996) Crystallographic Methods and Protocols .Humana Press.
-Köning,P.(1993) The X-Ray structure of the GCN4-bZIP bound to ATF/CREB site DNA shows the complex depends on DNA flexibility.J.Mol.Biol. , 233, 139-154.
-Panagiotidis,CA.(1995) Polyamines alter sequence-specific DNA-protein interactions. Nucleic Acids Research, 23, No 10, 1800-1809.
-Phillips,D.M.P.(1971) Histones and Nucleohistones. Plenum Press.
-Porschke,D.(1991) Nature of protamine-DNA complexes.J.Mol.Biol., 222, 423-433.
-Saenger,W.(1987) DNA-ligand interactions.From drugs to proteins. Plenum Press.
-Sinden,R.(1994) DNA Structure and Function. Academic Press.
-Subirana,JA.(1990) Analysis of the charge distribution in the C-terminal region of histone H1 as related to its interaction with DNA.Bioplymers, 29, 1351-1357.
-Suzuki,M.(1994) A frame-work for the DNA-protein recognition code of the probe helix in transcription factors: the chemical and stereochemical rules.Structure, Vol 2,
No 4, 317-326.
-Voet & Voet.(1995) Biochemistry. John Wiley & Sons INC.
-Vogel, H.(1977) Nucleic Acid-protein recognition. Academic Press.
-Vasilescu,D.(1990)Water and Ions in Biomolecular Systems. Birkhöuser Verlag.
-Wolffe,A.(1996) Chromatin, structure and fucntion. Academic Press.
-Zhang,W.(1996) Large electrostatics differences in the binding thermodynamics of a cationic peptide to oligomeric and polymeric DNA.Proc.Natl.Acad.Sci., 93, 2511-2516.
{kind=link}
{kind=link}
{kind=link}