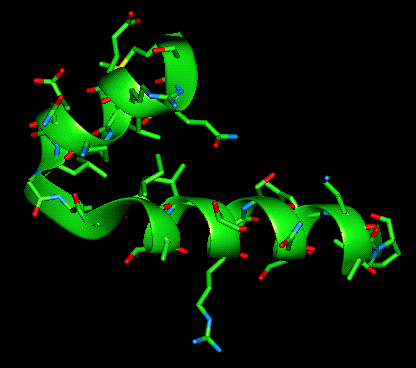
Helix-turn-helix motif (with and without amino acid sidechains shown)
In eukaryotic systems, there are other DNA-binding motifs which occur such as the zinc finger motif or the leucine zipper but these shall not be discussed further.
Although the DNA-binding motif is contained within one polypeptide chain, many DNA-binding proteins bind as dimers. This dimerization serves to orient the two molecules optimally for DNA binding. It is the second of the two helices in this motif which is termed the recognition helix as it binds in the major groove of B-form DNA, its amino acids close enough to the operator DNA to make contact.
The dimer is constructed such that it has bifold symmetry allowing the recognition helix of the second protein sub-unit to make the same groove binding interactions as the first. The distance between the recognition helices is 34 angstroms which corresponds to one turn of the B-DNA double helix. This means that when the recognition helix of one sub-unit binds in the groove of a specific region of DNA, the second sub-units' helix can also bind in the DNA groove, one turn along from the first helix. This is why the recognition helix is so called.
Because one turn separates the recognition helices, the central portion of the protein dimer faces the narrow groove of the DNA double helix. As a result of this, there are no DNA-protein interactions in this area.
The recognition alpha-helices are oriented within the dimer such that each is aligned with the major groove of the DNA, allowing the base pairs to make contact with the recognition helix.
The side chains of the recognition alpha-helices extend towards the base pairs within the major groove and so are able to interact with their edges. In addition to these, the protein sub-unit makes a considerable number of interactions along the sugar-phosphate backbone of the DNA on either side of the major groove.
Although the DNA in this complex adopts the B-form, it is far from the classical description of this conformation. The binding of the protein dimer causes a large number of distortions to the regular twist of the double helix. The central section of DNA, which as already mentioned makes no interactions with the protein dimer, is twisted more than usual and is referred to as being 'overwound'. The ends of the DNA suffer the opposite of this so their twist is reduced or 'underwound'. Whereas the classical form of B-DNA is linear, the DNA in one of these protein-DNA complexes has a distorted axis, one which curves towards the protein dimers' recognition helices. This has the effect of making the minor groove more narrow in the central portion which faces the dimer and wider at the ends.
In spite of all the observed protein-DNA binding interactions, the affinity of the DNA for protein appears to be mediated by the structural properties of the DNA double helix rather than by any interactions unique to that sequence. That is to say, the ability to tolerate certain structural distortions from the classical B-form determines how well the DNA will bind the protein dimer. Not only is the affinity of the protein for the DNA mediated by these non-sequence specific interactions but the differential affinities of repressors and activators for different operator DNA regions also. The non-specific DNA-protein interactions involving the sugar-phosphate backbone contribute to these structural changes and in these regions, there are many hydrogen bonds formed between these phosphate groups of the DNA and the NH groups of the protein backbone.
DNA conformation stabilising interactions are found along most of the polypeptide chain involving many amino acid residues and additional protein-protein sub-unit interactions orient the recognition helices in the correct way. It is fair to say that the whole protein contributes to the affinity and specificity of the DNA-protein complex.
Considered as the monomeric sub-unit, it is clear that no hydrophobic core exists which implies that a monomer would not be stable enough to exist in isolation. As explained in general terms above, two protein sub-units fit together to form a hydrophobic core which orients the two recognition helices.
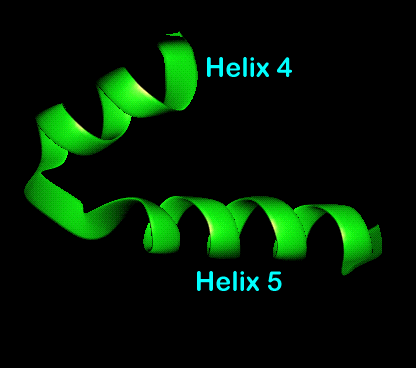
The N-terminal helices of each sub-unit contact each other as well as the core of the dimer so forming a stabilising globular core which was not possible when in the monomeric form. After these helices comes the third which is relatively long and fulfils the role of structural link between the N-terminal region or core and the recognition helices. Helices 4,5 and 6 form a "head" containing the recognition helix and helix-turn-helix motif. Helices 4 and 5 represent the helix-turn-helix motif with helix 5 being the recognition helix for DNA binding.
L-Tryptophan binds to the repressor protein in a site present in each monomeric protein between the long third helix and recognition helix five. It is for this reason that the dimer binds two molecules of l-tryptophan. When this tryptophan binding occurs, the recognition helices (no 5 in each sub-unit) are reoriented relative to the inactive form. The effect of the bound l-tryptophan molecules is to orient the recognition helices correctly for DNA binding. The helices are moved to 34 angstroms relative to each other to match the periodicity of the DNA twist.
When l-tryptophan is lost from the complex, the recognition helices tilt inwards to remove the empty cavity and this 5-6 angstrom shift prevents the binding of the repressor to the DNA sequence. The bound conformation can be mimicked in the absence of l-tryptophan by using a point-mutated protein. Residue 77 of alanine and the side chain points into the cavity. If this residue is changed to a bulkier amino acid like valine, the active conformation is maintained.
Between each protein monomer sub-unit and the operator DNA sequence are 14 hydrogen bonds. Only two of these involve the functional groups of a base, the others consisting of the interactions between the unesterified oxygens of six phosphate groups. The two functional group mediated interactions are between arginine 69 and guanine -9.
Four of the 14 hydrogen bonded direct contacts between the half-repressor and the operator involve amino acids outside of the helix-turn-helix motif. There are six additional contacts between the protein monomer and the operator which involve solvent, three each to the base pairs and to the phosphates. Three of the water molecules that are involved in hydrogen bonding between the half-repressor and half-operator are well ordered. Two of these bridge the amide nitrogen atoms of isoleucine 79 and alanine 80 to the hydrogen bond acceptors of adenine 5 and guanine 6. The water molecule that bridges isoleucine 79 to N7 of guanine 6 also appears to possess an additional interaction with the sidechain nitrogen of lysine 72. The third bridges the sidechain hydroxyl of threonine 83 to N6 and N7 of adenine 7. Two water molecules form hydrogen bonds between phosphate 3 and the sidechain oxygens of threonine 81 and threonine 53. In addition to these two water mediated hydrogen bonds, phosphate 3 also forms hydrogen bonds to arginine 54 and the indole nitrogen of a bound tryptophan molecule. The last solvent mediated contact involves a calcium ion which bridges phosphate 1 of the repressor to the sidechain of aspartic acid 46.
The fourth amino acid within the turn of the helix-turn-helix motif, glycine 78, is close enough to the hydrophobic surface of thymine 4 and adenine 5 of the operator to exclude water. This surface is hydrophobic no matter what the DNA sequence is and cannot discriminate against repressor sequences because other bases can be accommodated without causing steric clashes. This explains why amino acids other than glycine at position 78 disrupt the binding between the operator and repressor.
It has been shown that the main effect of the corepressor, l-tryptophan, is to orient the reading heads such that the helix-turn-helix motif can penetrate successive turns of the major groove of the operator1. The residues of the l-tryptophan binding pocket, as well as the corepressor itself, interact with the operator directly, As already mentioned, the indole nitrogen of l-tryptophan forms a hydrogen bond to phosphate 3. The carboxylate group of the corepressor forms a hydrogen bond such that the guanidino sidechain of arginine 84 interacts with phosphate 2. The corepressor is the most firmly fixed unit in the complex.

The Trp repressor dimer
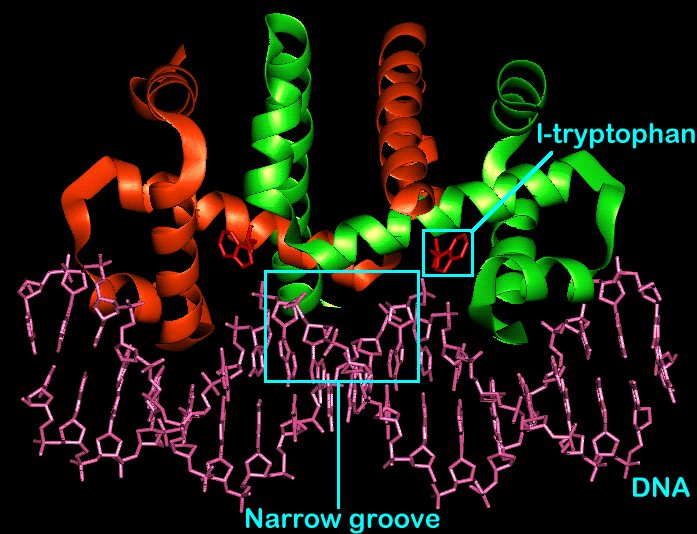
The Trp repressor/operator complex
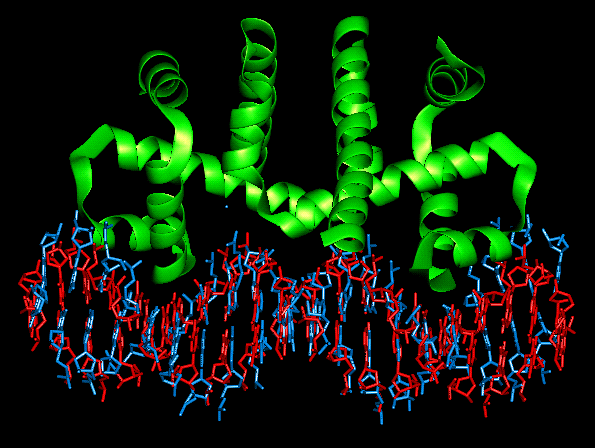
DNA in B-form:
classical
vs
distorted
The trp repressor/operator complex
The trp repressor is responsible for the control of l-tryptophan biosynthesis in E.Coli via a negative feedback loop. When no l-tryptophan is present, the operon is switched on so allowing the synthesis of l-tryptophan to proceed and rendering the repressor protein inactive. However, l-tryptophan binds to the repressor protein, so activating it to bind to the DNA operator and switch off the biosynthesis. This negative feedback process occurs as the concentration of l-tryptophan increases. The structural analysis of the trp repressor has shown that the 107 amino acids which compose it are arranged into 6-alpha helices as indicated below.
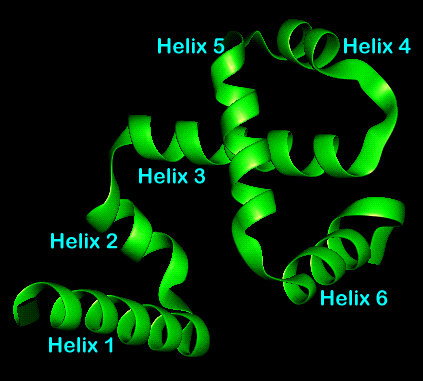
Repressor monomer with numbered helices

Repressor dimer showing hydrophobic core
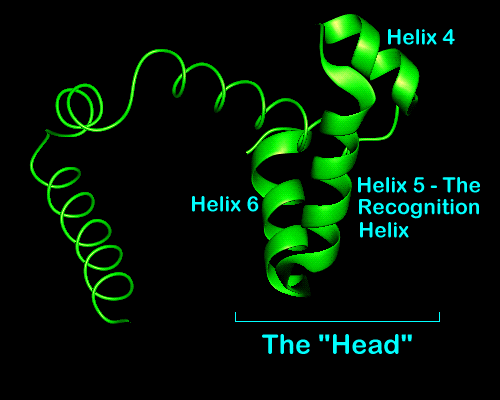
The repressor head with helices marked

Repressor dimer with bound l-tryptophan
Amino acid/base interactions
Within the complex, there are a number of interactions observed between the repressor and the operator. These vary from non-polar contacts to hydrogen bonds both with and without solvent molecules acting as bridges.
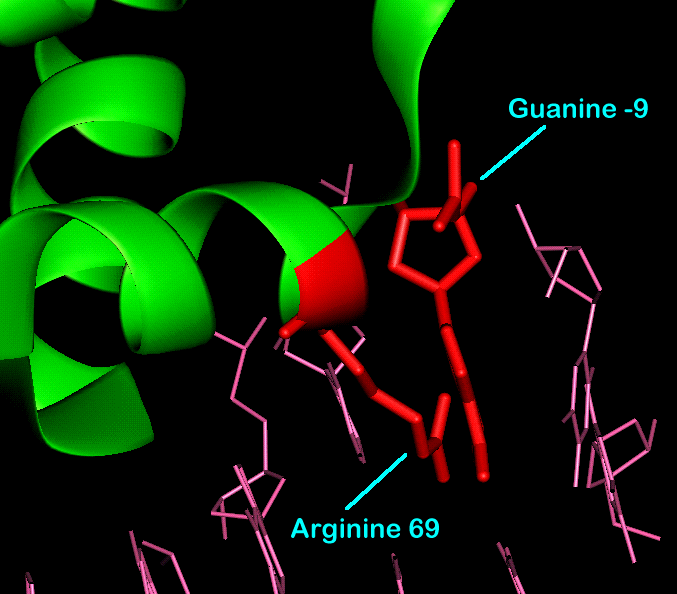
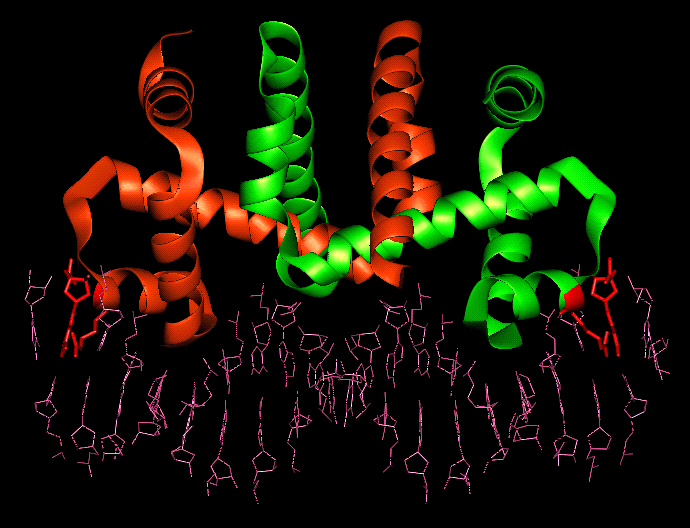
Functional group interaction between
Arginine 69 and Guanine -9
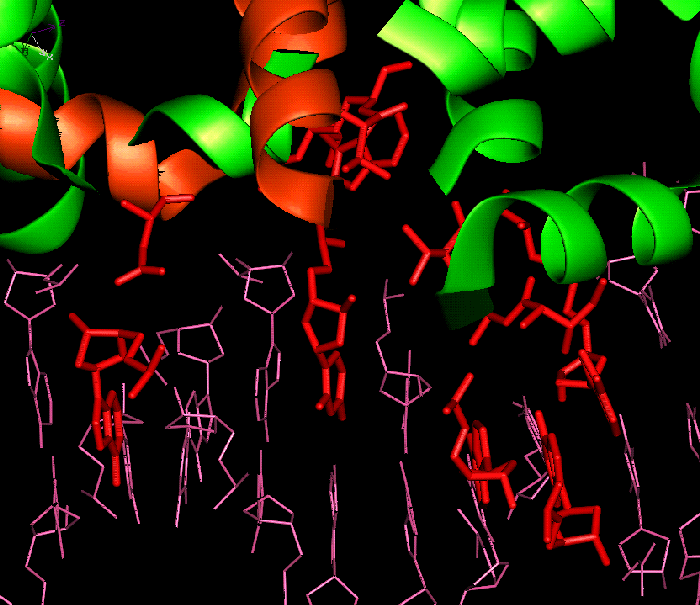
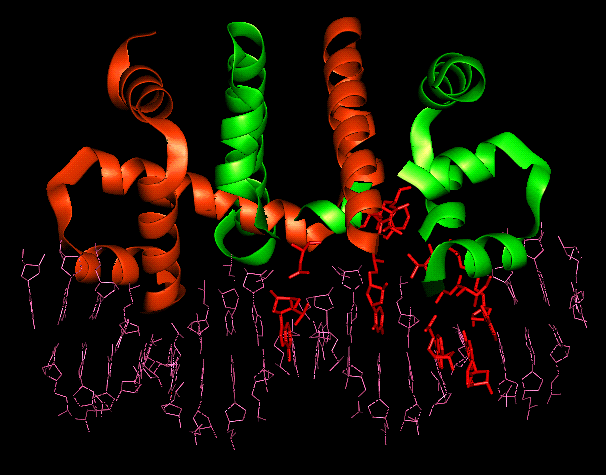
Bound dimer showing the
residues
involved in solvent bridged interactions

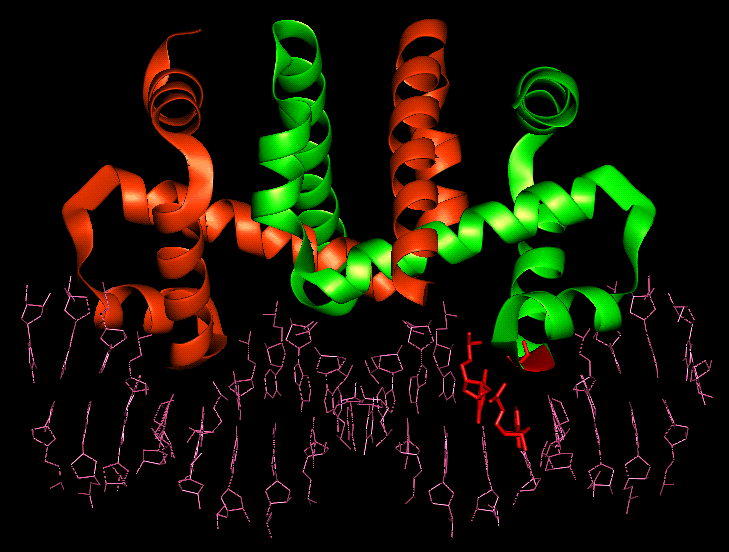
Bound dimer showing
Glycine 78, Thymine 4 and Adenine 5
Summary
All the information given above points to the following conclusion: the recognition system operating between the operator and repressor functions largely independently of any sequence specific interactions. Within the trp repressor/operator complex, there are no direct hydrogen bonds between the functional groups of the bases and the protein that can account for the specific affinity of the trp repressor for the operator sequence. There are also no non-polar contacts between the surface of the bases and the sidechains that could account for the specificity either. It appears that it is the propensity of the DNA sequence to adopt certain conformations and the protein's ability to stabilise these conformations that best explains the specificity observed in this and other protein-DNA complexes. It is also interesting to note the number of interactions that occur as a result of being solvent mediated. In spite of this, the complex is very stable as evidenced by the thermal parameters observed for the various parts of the complex. When the repressor is unbound, the sidechains on the surface are mobile as expected but on forming the complex, the temperature factors for the sidechain atoms that are in contact with the operator are equal to or less than those of the peptidic backbone.
References and Images