(c) O.S. Smart 1995, all rights reserved
This section is adapted from part 1.2.2. of O.S. Smart, "Simulation of a conformational rearrangement of the substrate in D-xylose Isomerase", University of London Ph.D. thesis (1991).
where R_i are the position vectors of the nuclei and the Z_i the charges. The electronic wavefunction and potential energy are given by:
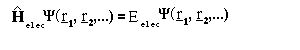
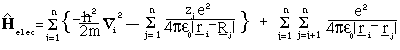
where r_i are the position vectors for the electrons.
In the case of the simplest molecule 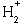 the exact solution
can be found numerically, but with some difficulty
(Wind, H., 1965, J. Chem. Phys. 42:2371-2373).
In more complicated systems, it is necessary to take a different
approach. The LCAO method, which is commonly used, expresses a
trial molecular orbital (MO) as a Linear Combination of the
individual Atomic Orbitals
the exact solution
can be found numerically, but with some difficulty
(Wind, H., 1965, J. Chem. Phys. 42:2371-2373).
In more complicated systems, it is necessary to take a different
approach. The LCAO method, which is commonly used, expresses a
trial molecular orbital (MO) as a Linear Combination of the
individual Atomic Orbitals 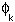 for the molecule:
for the molecule:
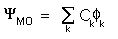
The variation principle can be exploited to find values for the
coefficients 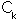 . The principle states that the expectation value
. The principle states that the expectation value
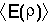 . of a
Hamiltonian
. of a
Hamiltonian  calculated with any function
calculated with any function
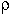 :
:
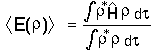
must lie above the true ground state energy (the integration is over all
space). Or put another way
if we have a trial wavefunction for the molecule and we make a
change which lowers the energy then the new function is a better
approximation to the true wavefunction to the system.
Thus, the optimal
value for the coefficients
can be found by adjusting the coefficients so as to minimize
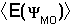 .
.
A further complication arises because of the last term of the Hamiltonian which describes electron-electron repulsion. Without this term it would be possible to use the method of separation of variables to solve the Schrödinger equation independently for each electron, but this is not possible when the term is taken into consideration. The standard approximation, developed by Hartree and Fock, to avoid this problem is to assume that each electron moves in the average field due to the nuclei and the other electrons. An initial set of wavefunctions is guessed for the molecule. In the LCAO approach a series of fixed phi called the basis set, is chosen (this must at least give a representation for each occupied atomic orbital of each atom) and the initial guess is a set of coefficients C_k (n.b. this must satisfy the Pauli principle and be antisymmetric - see Hinchliffe). The optimal coefficient for each atomic orbital in turn is found while keeping the other orbitals frozen. The procedure is continued until all the coefficients remain unaltered in a pass through the cycle and become self-consistent. The method is known as the self-consistent field approach (SCF).
This is commonly known as
or the advanced topic: The application of quantum chemistry to protein simulation. (also contains references for this section).