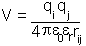
Back to main Molecular Forces index
Back to previous unit Covalent Interactions
On to next course unit The effect of solvent and hydrophobic interactions
As the name implies non-bonded interactions act between atoms which are not linked by covalent bonds. Like most things this is simple to state but can be confusing to apply in practice! In most approaches then atoms which are involved in a bond angle are also not regarded as having a non-bonded interaction. 1-4 interactions (those between the end atoms involved in a dihedral angle) are sometimes given an additional scaled down nonbonded interaction. Similarly the interaction between a metal ion and its liganding atoms is usually regarded as non-bonded and treated by the kind of approach set out here but are sometimes represented by the bond and bond angle terms of the potential energy function (e.g., in representing the iron atom in the haem group found in globins).
As mentioned in the introduction to this section electromagnetic interactions dominate on the molecular scale and provide the fundamental basis for all the different bonded and non-bonded interactions discussed here. This is clearest in the case of electrostatic interactions where charges on nuclei and electrons interact according to Coulomb's law:
where 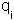 and
and 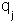 are the magnitude of the charges,
are the magnitude of the charges, 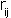 is their separation,
is their separation, 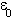 the permittivity of free space and
the permittivity of free space and 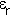 the relative dielectric constant of the medium in which the charges
are placed (if you do not remember this consult your high school
Physics textbook!). The strictly correct way to use the law would
be to consider every nucleus and electron separately, plug it
into the Schrödinger equation and apply quantum chemical
methods to solve the equation for the spatial configuration of
nuclei we are interested. As already mentioned this is completely
impractical for biomolecular systems. So instead we wish to develop
a useful model for the interactions between nuclear centres (commonly
called "atoms") without having to explicitly deal with
the electrons in a system.
the relative dielectric constant of the medium in which the charges
are placed (if you do not remember this consult your high school
Physics textbook!). The strictly correct way to use the law would
be to consider every nucleus and electron separately, plug it
into the Schrödinger equation and apply quantum chemical
methods to solve the equation for the spatial configuration of
nuclei we are interested. As already mentioned this is completely
impractical for biomolecular systems. So instead we wish to develop
a useful model for the interactions between nuclear centres (commonly
called "atoms") without having to explicitly deal with
the electrons in a system.
The simplest approach is to just to consider the formal charges of the protein. Formal charges show whether chemical groups are ionized i.e., whether an atom or set of atoms has lost or gained an electron. Isolated amino acids (in neutral solution) are zwitter ionic - this means that although the molecule has no overall charge it carries both a negatively charged group and a positively charged group:
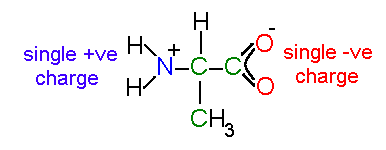
an isolated alanine residue in solution
Note that the atoms in a charged carboxylic acid are equivalent - the double bond is delocalized across the group.
In proteins the individual amino acids are polymerized (in a condensation reaction - releasing water). This results in a peptide backbone which is electrically neutral with the exceptions of the ends of the chain. In a normal protein the amino end carries a positive charge (-NH2+) and the carboxyl end carries a positive charge (-CO2-). In some cases (e.g., a number membrane polypeptides) the ends are chemically modified to avoid these charges (for instance by acetylation of the amino end group).
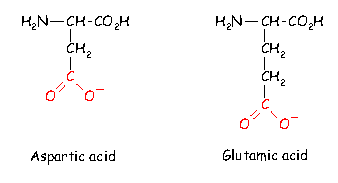
Most of the standard amino acids found in proteins have uncharged side chain groups. However, there are a number of basic residues which are positively charged at normal pH (if you do not know the meaning of the pH scale of acidity then consult any text book on physical chemistry) :
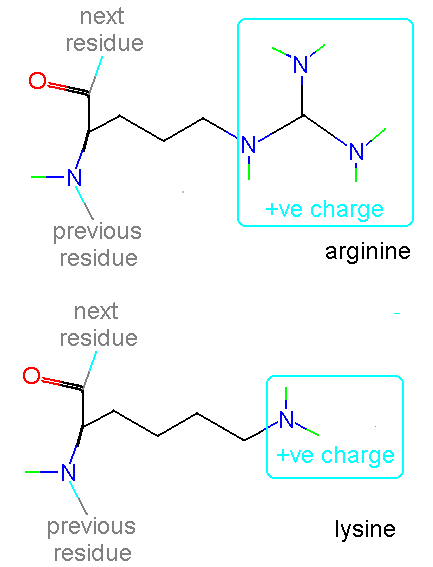
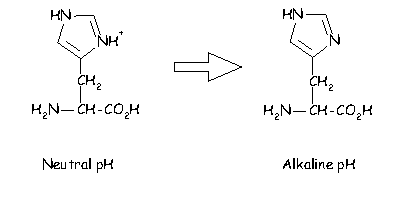
In addition histidine is normally charged at neutral pH (the charge normally residing on the delta carbon but sometimes on the epsilon). When the residue is placed in a basic environment it loses a proton and becomes uncharged:
Very many proteins bind inorganic ionic species such as metal ions - indeed such ions can play a crucial role in the mechanism of a protein. An example of this is the enzyme xylose isomerase which has two Mg2+ ions at the active site which coordinate to the substrate, polarizing it and stabilizing the transition state in the reaction.
As might be expected a positively charged lysine or arginine residue can form a strong interaction with a negatively charged asp or glu group. In proteins this interaction is referred to as a salt bridge.
Click here if
you would like to see an example of a salt bridge.
if
you would like to see an example of a salt bridge.
In practice salt bridges are relatively rare in proteins and in practice they normally occur on the surface as opposed to internally. An exception is when an internal salt bridge is involved in the catalytic mechanism of an enzyme such as in the asp-his-ser triad of serine proteases (a classic example of the structural basis of enzyme activity).
The reason for this is that although an internal salt bridge is a strong interaction in comparison to having the isolated residues widely separated in a vacuum it is normally destabilizing for a protein. This apparent paradox is due to that fact that when considering the effect of an interaction one must consider the difference in the (free) energy between the folded and unfolded but solvated states. In the unfolded state the residues involved in a salt bridge would be widely separated but each making very favourable interactions with water molecules (for advanced students - there is an entropic contribution to this). These interactions are lost when the same residues are buried in the largely hydrophobic core of the protein. Similar arguments apply to practically all considerations of elucidation of the energetic contributions to protein folding or ligand binding - normally a small overall free energy advantage arises from the balance between large but cancelling contributions. This is one of the problems which make the computation prediction and analysis of protein behaviour so difficult.
The electrostatic interactions between groups which carry no formal overall electrical charge are of fundamental importance to biomolecular structure. The source of these is that uncharged species can still have a large inherent polarization - the orbitals around the molecule are distributed in such a way that parts of the molecule have less electrons and thus carry a positive charge and other parts have an excess and are therefore negatively charged. Some atoms (O, N and to a lesser extent S) have a tendency to attract electrons (filling the valence shells) and are termed electronegative. Others (notably metallic atoms) have a tendency to lose electrons. In extreme this tendency causes one atom completely to lose an electron to another - leading to the formation of formally charged species or ions (see previous section). In less extreme conditions electrons are shared between two atoms in a covalent bond but are pulled towards one partner. The classical example of this is the water molecule:
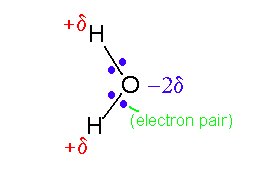
As oxygen is electronegative it draws the electrons in the bonds it shares with the hydrogen atoms towards it. The hydrogen atoms are left with a net positive charge and the oxygen is negative. In the case of a water molecule the value for the effective charge on each hydrogen atom is quite large - around one third of an electron. Together with the short distance between the oxygen and hydrogen atoms this results in the water molecule having a large dipole moment. Two water molecules can therefore form a strong electrostatic interaction:
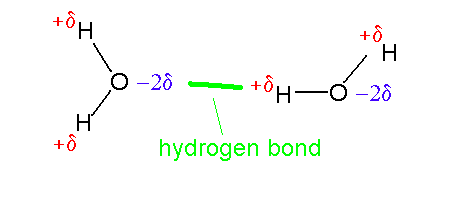
This interaction is known as a hydrogen bond (incidental note the lowest energy water dimer is not flat there is an angle of around 23 degrees between the planes of the molecule). The bond is normally around 2.8Å long (measured from oxygen to oxygen). This length results from the interplay between the electrostatic stabilizing factor and the repulsion between the oxygen atoms as they come closer. Water molecules can form a network of hydrogen bonds. The fact that it is strong has a number of important consequences:
 click to see larger picture
click to see larger picture
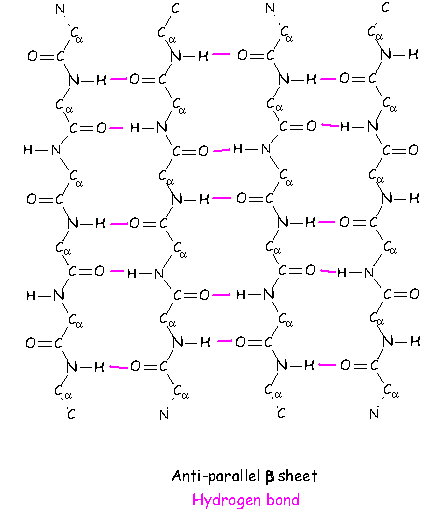 click to see larger picture
click to see larger picture
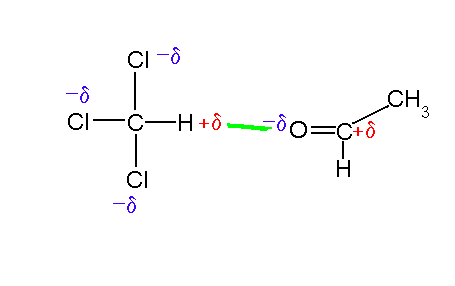
Many side chain groups in proteins can form hydrogen bonds:
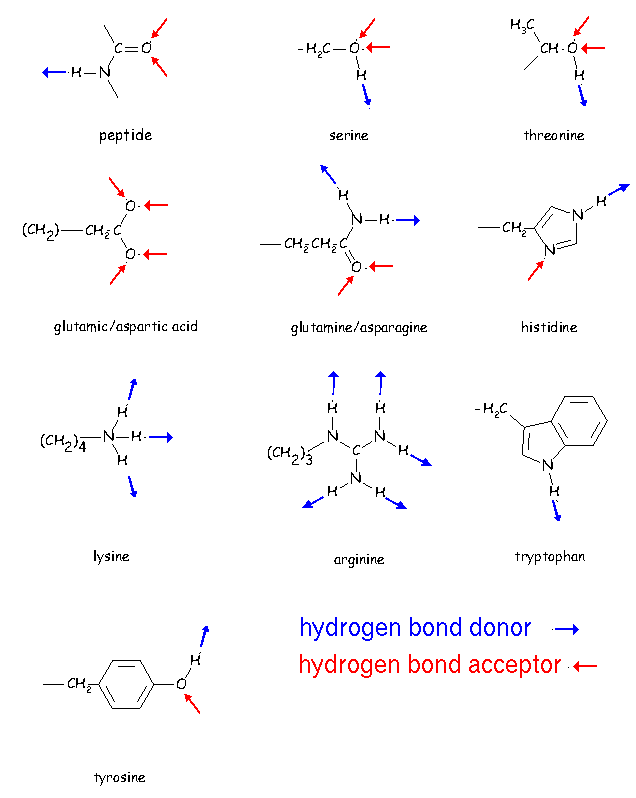 click to see larger picture
click to see larger picture
In addition interactions can be formed between groups carrying a formal charge and hydrogen bonding atoms e.g.,
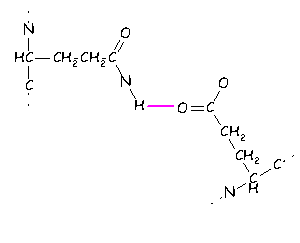
This is normally regarded as an especially strong variant of the hydrogen bond.
We have seen that electrostatic interactions are of fundamental importance to proteins. We shall now briefly examine the manner in which they are normally treated in computational studies.
The most common approach is to place a partial charge at each atomic centre (nucleus). These charges then interact by Coulomb's Law. The charge can take fractions of an electron and can be positive or negative. Charges on adjacent atoms (joined by one or two covalent) bonds are normally made invisible to one another - the interactions between these atoms being dealt with by covalent interactions. Note that the concept of a partial charge is only a convenient abstraction of reality. In practice many electrons and nuclei come together to form a molecule - partial charges give a crude representation of what a neighbouring atom will on average "see" due to this collection.
The standard modern way to calculate partial charges is to perform a (reasonably high level) quantum chemical calculation for a small molecule which is representative of the group of interest (e.g., phenol is considered for tyrosine). The electrostatic potential is then calculated from the orbitals obtained for many points on the molecular surface. A least squares fitting procedure is then used to produce a set of partial charges which produce potential values most consistent with the quantum calculations. (Cieplak, P., Cornell, W.D., Bayly, C., Kollman, P.A. (1995) application of the multimolecule and multiconformational RESP methodology to biopolymers - charge derivation for DNA, RNA and proteins. J. Comp. Chem. 16:1357-1377). Older procedures used methods in which orbital populations are simply split between atoms (Mulliken Population Analysis). All though much simpler these charges do not produce a reasonable representation of the electrostatic potential around a molecule - which is usually what is of interest in a simulation.
To see the kind of values typical for partial charges look at
picture of a salt bridge .
As mentioned previously using partial charges at nuclear centres is the crudest effective abstraction. To obtain a more accurate representation two approaches are common. The first is to add dipole, quadrapole and higher moments to the nuclear centres (do not worry if these terms are unfamiliar, see: Stone, A.J., Price, S.L. (1988) Some ideas in the theory of intermolecular forces: Anisotropic atom-atom potentials. J. Phys. Chem. 92:3325-3335). The second is to introduce further non-nuclear centres - this is commonly done to represent the anisotropy in potential cause by lone pairs on oxygen atoms (Cieplak, P., Cornell, W.D., Bayly, C., Kollman, P.A. (1995) application of the multimolecule and multiconformational RESP methodology to biopolymers - charge derivation for DNA, RNA and proteins. J. Comp. Chem. 16:1357-1377).
In many respects electrostatic interactions provided the biggest problems to computational studies of protein behaviour. By their nature they are long range and dependent on the properties of the surrounding medium (see discussion of dielectric effects). A simple rule of thumb is that the more highly charged a system the harder it is to simulate - thus simulations of liquid argon can do a wonderful job, hydrocarbons are fairly easy, water becomes difficult and proteins more so. The limit is reached with nucleic acids like DNA which are aqueous complex salts (each base having a charge of minus two) with counter ions and solvent having important effects on structures. Usually some sort of "fudge" has to be made in simulations to keep DNA stable at all!
The normal treatment for partial charges is to assume they are fixed. In practice the electric field caused by other atoms and molecules will polarize an atom effecting its electron distribution and thus its partial charge. In turn the partial charge produces an electric field which affects neighbouring charges and thus fields. To be able to work out the partial charges a self consistency cycle is normally used. The process of polarization has an energetic effect. In practice it is difficult to find adequate parameters to treat systems as complex as proteins (work has recently been concentrating on systems such as sodium ions in water). Induction effects can be shown to decay by a r-6 relations so they can normally be regarded as implicitly corrected for when the dispersion term is fitted as discussed in the next section.
You will probably be familiar that at low temperatures gases such as argon liquefy. The attractive interactions which cause this are called dispersion. Although they also occur between charged atoms they are usually overwhelmed by the stronger electrostatic terms and so are normally only of importance for uncharged groups. To really understand dispersion effects one must turn to 2nd order perturbation theory in quantum mechanics. You will probably be happy to know that we shall not be doing this! Instead a simple physical picture will be given.
Imagine that we have an atom of argon. It can be considered to
be like a large spherical jelly with a golf ball embedded at the
centre. The golf ball is the nucleus carrying a large positive
charge and the jelly represents the clouds of electrons whizzing
about this. At a point external to the atom the net average field
will be zero because the positively-charged nucleus' field will
be exactly balanced by the electron clouds:

However, atoms vibrate (even at 0K) and so that at any instant
the cloud is likely to be slightly off centre. This disparity
creates an "instantaneous dipole":
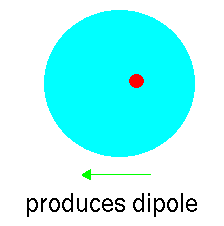
Suppose that we have another argon atom close to the first. This
atom will see the electric field resulting from the instantaneous
dipole. This field will effect the jelly inducing a dipole:
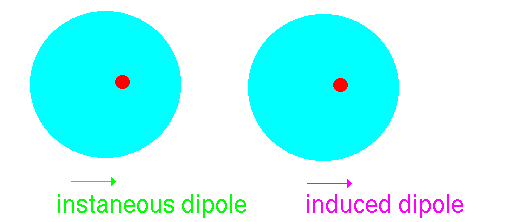
The two dipoles attract one another - producing an attractive
interaction.
The Dispersion interaction can be shown to vary according to the inverse sixth power of the distance between the two atoms:
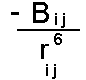
depends on the nature of the pair of atoms interacting (in particular their polarizability). It is normal to parameterize the dispersion empirically using structural and energetic data from crystals of small molecules. It is not possible to use simple quantum chemical calculations to find parameters. This is because most quantum chemical calculations use the self consistent field approximation (SCF). In this each electron is solved independently keeping the other orbitals frozen (in a self consistency). This effectively means that electrons only experience a time averaged picture of other electrons - so that dispersion cannot come into effect. More advanced methods in quantum chemistry introduce methods to tackle "electron correlation" to avoid this problem.
When two atoms are brought increasing close together there is a large energetic cost as the orbitals start to overlap. In the limit that the atomic nuclei where coincident the electrons of the two atoms would have to share the same orbital system. The Pauli exclusion principle states that no two electrons can share the same state so that in effect half the electrons of the system would have to go into orbitals with an energy higher than the valence state. For this reason the repulsive core is sometimes termed a "Pauli exclusion interaction".
The simplest and oldest way to represent the repulsive core for atoms is by using a "hard sphere" model. In this atoms have a characteristic radius (below the van der Waals radius) and cannot overlap. This approach can be adopted computationally but is more commonly seen in physical models using plastic spheres such as CPK:

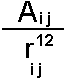
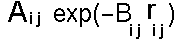
The dispersion and repulsion terms discussed above are commonly grouped together into the Lennard-Jones or 6-12 potential:
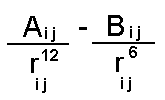
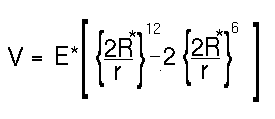
atom type van der Waals radius van der Waals well depth
in Å in kcal/mol
C (aliphatic) 1.85 0.12 O 1.60 0.20 H 1.00 0.02 N 1.75 0.16 P 2.10 0.20 S 2.00 0.20
It is important to note that the Lennard-Jones interaction between uncharged atoms (such as CH3 groups) is less attractive than that between charged groups such as oxygens. The difference is that the contribution from electrostatics will dominate the L-J interactions.
In cases where uncharged groups form compact structures van der Waals energies are often cited as stabilizing the conformation. Although partly true very often the major contribution comes rather from hydrophobic exclusion.
On to next course unit The effect of solvent and hydrophobic interactions