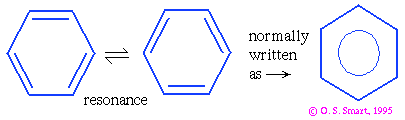 (Click on icon to proper sized image).
(Click on icon to proper sized image).
Back to main PPS course Index
Back to main Molecular Forces index
Back to previous unit Preliminary Considerations
On to next course unit Non-bonded Interactions
We have reviewed the methods of representing molecules with quantum chemical methods and found that (with some limited exceptions) it is impractical to apply these methods to biomolecules. It is therefore necessary to consider a more simple level of representation where "atom" centres are considered at the nuclear position for each atom and these interact by a variety of effects. The models developed should both aid qualitative understanding of the physical basis of various experimental effects and provide a quantitative insight via their implementation as part of a potential energy function. For convenience we divide the topic into consideration of covalent interactions (namely bonds, bond angles and dihedral angles) and non-bonded interactions (electrostatic interactions, induction and dispersion effects, repulsion terms) which are treated in the next section.
In simple terms a covalent bond exists between two atoms if they share electrons between them. In contrast, an ionic bond is formed if electrons are transferred between atoms (e.g., in sodium chloride an electron is given up by the sodium atom to form a Na+ ion and accepted by the chlorine atom to form a chloride, Cl- ion) - such formally charged interactions will be dealt with later). A single bond is formed when one pair of electrons is involved and a double bond when two pairs are involved. In quantum chemical terms such a picture is overly simplistic as the although a bonding orbital results in an increase in electron density between the atoms it also spreads over the rest of the molecule. This is particularly in the case of delocalized bonding. The "classical" example of delocalized bonding is the benzene molecule - which can be described as resonance hybrid between a number of alternate structures:
(Click on icon to proper sized image).
If you are unfamiliar with this then look at any high school chemistry text book or even better have a look under "benzene" and "resonance" in Atkins. Delocalized bonding is important in protein structure: it is why the peptide bond is planar (in Section 3) and it occurs in phenylalanine, tryptophan, glutamic acid, arginine side chains.
The standard way to approximate the potential energy for a bond
in a protein and most other molecules is to use a Hooke's law
term: 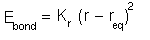
where r is the length of the bond (i.e., the distance between the two nuclei of the atoms between which the bond acts), r_eq is the equilibrium bond length and K_r is a spring constant. This basically represents the bond as a spring linking the two atoms.
A few points can be made about this representation:
Typical values for bond constants and equilibrium bond lengths taken from the AMBER potential energy function:
Atom pair r_eq in Å K_r in kcal/(molÅ2)
C = O 1.229 570 C - C2 1.522 317 C - N 1.335 490 C2 - N 1.449 337 N - H 1.010 434
These are the bond parameters necessary in representing a glycine residue and its connections to neighbouring residues. The atom types are:
O a carbonyl oxygen C a sp2 carbon (such as that attached to an O) N a main chain nitrogen atom H a hydrogen atom attached to the N C2 a "united atom"group
(united atom means that instead of representing the carbon atom and the two hydrogen atoms which are bonded to it separately only one centre is considered - further explanation and picture)
Small molecule X-ray crystal structures are typically used to obtain r_eq values. The spring constants K_r are found by performing normal mode calculations and comparing the results with experimental microwave frequencies (as done in the case of AMBER). Parameters can also be obtained from ab initio quantum chemical calculations.
Click here if you would like to see a graph of the energy for a typical bond.
if you would like to see a graph of the energy for a typical bond.
A bond angle  between
atoms A-B-C is defined as the angle between the bonds A-B and
B-C:
between
atoms A-B-C is defined as the angle between the bonds A-B and
B-C:
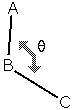
As bond angles are found (experimentally and theoretically) to
vary around a single value it is sufficient in most applications
to use a harmonic representation (in a similar manner to the bond
potential): 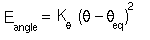
Typical values for equilibrium bond angles and bond angle constants taken from the AMBER potential energy function:
Anglein degrees
in kcal/(mol.degrees2)
C-N-H 119.8 35.0 C2-N-C 121.9 50.0 C2-N-H 118.4 38.0 C-C2-N 110.3 80.0 C2-C-O 120.4 80.0 C2-C-N 116.6 70.0 O-C-N 122.9 80.0 (explanation of atom types)
These are the bond angle parameters necessary in representing a glycine residue and its connections to neighbouring residues. A bond angle around 109 degrees means that the central atom is tetrahedral (with four other atoms bonded to it):

The angle around the C beta atom of an alanine residue - showing the three hydrogen atoms bonded to the carbon. In contrast an angle around 120 degrees indicates a flat (sp2) central atom with three other atoms bounded to it:
 This shows the angles made
around a main chain nitrogen atom are all approximately equal to 120 degrees:
consequently the group is planar.
This shows the angles made
around a main chain nitrogen atom are all approximately equal to 120 degrees:
consequently the group is planar.
The source of bond angle parameters is the same as for bonds: high resolution small molecule X-ray structures for eqilibrium values and either spectroscopic data or ab initio calculations for force constants.
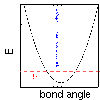 if you would like to see a graph of the energy for a typical bond angle.
if you would like to see a graph of the energy for a typical bond angle.
Dihedral angles
In case you are not too familiar with the what a dihedral angle is
why not revise the Peptide Torsion Angles section that you saw earlier in the
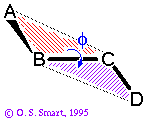
course. Formally the dihedral angle  (also known as a torsion angle)
between four atoms A-B-C-D is defined as the angle between the
the planes ABC (marked by red lines) and
BCD (marked in purple):
(also known as a torsion angle)
between four atoms A-B-C-D is defined as the angle between the
the planes ABC (marked by red lines) and
BCD (marked in purple):
Thus a dihedral angle of zero is a cis conformation and 180 degrees is a trans conformation:
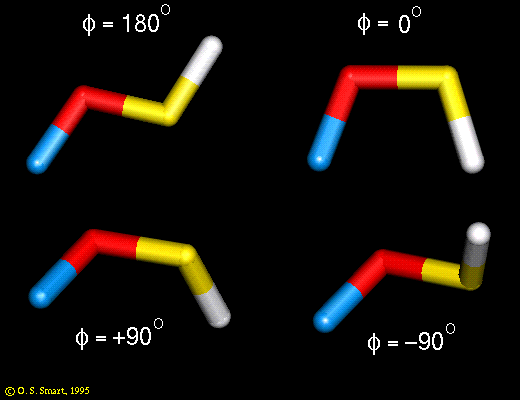 (Click on icon to proper sized image).
(Click on icon to proper sized image).
The standard functional form for representing the potential energy for a torsional rotation was introduced by Pitzer (Disc. Faraday Soc. 107:4519-4529, 1951):
![E_dihe= sum_(i=1)<sup>n</sup> (V_n/2)*[1+cos{n.phi-gamma}]](os_dihe.gif)
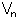 gives the energy barrier to rotation, n the number of maxima (or minima) in one full rotation and
gives the energy barrier to rotation, n the number of maxima (or minima) in one full rotation and
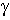 determines the
angular offset. The use of the sum allows for complex angular variation of
the potential energy (in effect a truncated fourier series is used).
Barriers for dihedral angle rotation can be attributed
to the exchange interaction of electrons in adjacent bonds (see Pauling).
Steric effects can also be important (see next section).
determines the
angular offset. The use of the sum allows for complex angular variation of
the potential energy (in effect a truncated fourier series is used).
Barriers for dihedral angle rotation can be attributed
to the exchange interaction of electrons in adjacent bonds (see Pauling).
Steric effects can also be important (see next section).
Click here 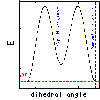 if you would like to see a graph of the potential energy terms used by the
AMBER
PEF to keep the peptide bond planar.
if you would like to see a graph of the potential energy terms used by the
AMBER
PEF to keep the peptide bond planar.