![]() Index to Course Material
Index to Course Material
![]() Index to Section 10
Index to Section 10
![]() Domains Front Page
Domains Front Page
![]() DIAL
DIAL
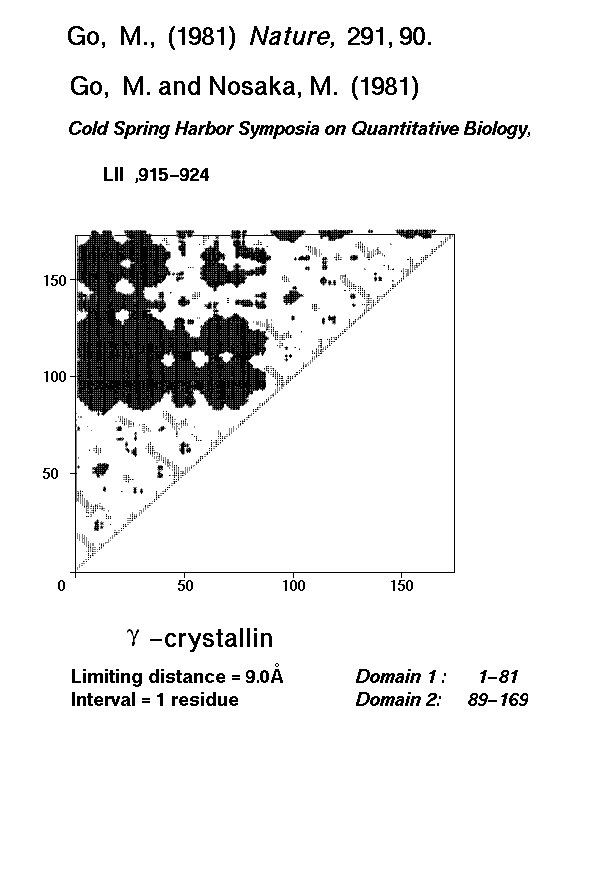
Go (1981) also exploited the fact that inter-domain distances are normally larger than intra-domain distances; all possible C-alpha - C-alpha distances were represented as diagonal plots (Go and Nosaka, 1987) in which there were distinct patterns for helices, extended strands and combinations of secondary structures. It is possible to identify even discontinuous domains using these distance diagonal plots. However, both methods assumed that domains comprise an ideal compact structure of amino acid residues and this is not universal.
Rose (1979) considered a protein molecule as a rigid body and defined three mutually perpendicular axes passing through the centroid. The domain disclosing plane (defined by the larger two axes) and the cutting line (corresponding to the third axis) were employed to identify continuous chain segments which corresponded to compact domains. By measuring the entire protein volume and the volume of dissected segments, two segments A and B of a continuous polypeptide AB were said to be domains if they fell on either side of the domain disclosing plane. An error function was applied to every pair of polypeptide segments of the protein.
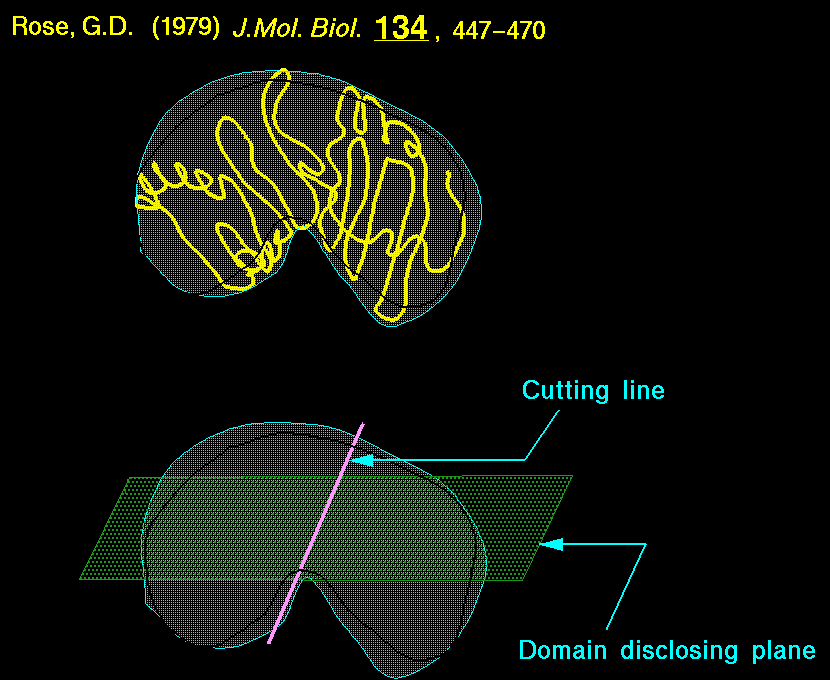
An important prerequisite for the error function was the input of rough boundaries of the domains. This method works strictly only for continuous domains and may identify several substructures as individual domains.
A binary clustering algorithm (Crippen, 1978) considered proteins as several small segments which need not be the secondary structural components of the protein. The initial segments were clustered one after another based on inter-segment distances. Segments with the lowest values were clustered and considered as a single segment thereafter. The stepwise clustering finally included the full protein.
Zehfus and Rose (1986) calculated compactness of substructures using solvent accessibility. Although this method can be extended to identify discontinuous domains (Zehfus, 1994), it is computationally expensive.
Argos (1990) analysed protein domains in the context of the composition and conformation of domain linkers. A graphical inspection was used in order to identify domains and their boundaries. Holm and Sander (1994) used a contact matrix to group residues in a protein in order to identify domains. Islam et al. (1994) also use contact matrices in order to identify domains in proteins.
Swindells (1995) have developed a method for the identification of domains in protein structures based on the idea that domains have a hydrophobic interior. This method is an extension of the procedure that recognises hydrophobic cores in a protein. The present method may fail for cases where domains do not have ideal hydrophobic cores.
The procedure of Sowdhamini and Blundell (1995), coded as a computer program DIAL,
As the procedure does not consider the protein as a continuous chain of amino acids there are no problems in treating discontinous domains. Special care is required, however, to identify situations where a secondary structure is shared between two domains (as in Catabolite gene activator protein, or glutathione reductase; refer to next page).
![]() Download the Catabolite gene activator
protein structure from your nearest PDB mirror: 3gap (302Kb)
[Bbk|BNL|ExP|Waw|Hal]
Download the Catabolite gene activator
protein structure from your nearest PDB mirror: 3gap (302Kb)
[Bbk|BNL|ExP|Waw|Hal]
This script illustrates the two domains and the alpha helix (helix 3 in the PDB file) which is shared by them.
Note the difference between this and:
Last updated 13th Jun '96